Volta cell (w/acid electrolyte) showing self discharge
H+ anode current
What is a battery?
What I learned
Ionic
chemistry
Single
electrode reactions
One
electrode 'battery' --- Zinc dissolving in acid
Single
zinc electrode circuit model
Zinc
in copper sulfate solution
Battery definitions
How
do batteries make chemical power available to do work?
Heat
or work
Preferred
current path is external
Introduction
Battery families
Zinc family overview
Volta
(1800) -- Zinc and copper in sulfuric acid --- single reaction battery
Volta's
flaw and Daniell's fix
Daniell
cell --- Much improved Volta battery with two electrolytes
Gravity
(Daniell type) cell
Silver
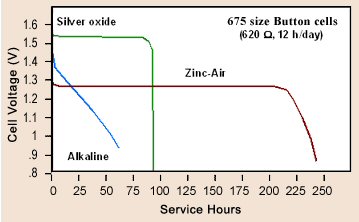
oxide battery -- basic electroyte with OH- ionic current
Zinc-air
(zinc-oxygen) battery
Weight
vs Ahr
Zinc-copper
alkaline battery
Zinc-carbon
(flashlight) battery
Rechargable batteries
Lead-acid
battery -- Unique single electrolyte battery with high voltage
Lithium
ion battery
High Power batteries
MIT
Professor Sadoway's liquid metal battery
Sodium
sulfur high power battery
Sodium
nickel-chloride medium power battery
Thermal
batteries
Electric
car batteries
Battery related
Cathodic
protection of ships
Electrochemistry
cell
Mysteries
High
electrolyte potential mystery
Zinc
sulfur mystery
Why
are zinc and copper so different?
Where
is the anode?
Chemistry Appendix
Metals
and non-metals in periodic table
Electrochemistry
basics
Sulfuric
acid as an electrolyte
Univ
of Sydney electrochemistry lecture
Appendix II
Measuring
standard reduction potential
Ionic
bonding
Ion
size
Element
reduction potentials
How do batteries really (really) work? I always wanted to know, so I spend three months reading and learning, and this essay is the result.
Units
1 eV = 96.5
kj/mol
F =
96.5 x 10^3 coulomb = [6.02 x 10^23 x 1.60 x 10^-19 coulomb]
Faraday (F) is the charge of a mole of electrons
Window into redox reactions
The cool thing about
batteries is that they open a window into chemisty. The theoretical, abstract
concept of reduction-oxidation (redox) reactions becomes real and
visible with batteries. The (net) voltage the electrons move through during
redox reactions, as they jump between atoms, becomes the battery voltage
measurable with a simple voltmeter.
What is a battery?
A battery
is a clever invention, almost magical, in that it releases chemical energy
as usable electrical energy. In standard exothermic chemical reactions
heat
is released as electrons 'jump' between atoms in the process falling through
a voltage difference (V) generated by the E fields of the atoms, typically
a fraction of a volt to 3.6V, each electron releasing (eV) joules of energy.
The cleverness of the battery design is that as electrons detach from of
one set of atoms (at the anode), their preferred path is to flow externally
through a load (doing work) as they drop through the voltage difference
(V), and upon arriving at the cathode they attach to a different set of
atoms. The E field voltage difference (V) between the electron orbitals
of the atoms releasing the electrons and those atoms accepting them is
coupled to the battery terminals making it visible outside
the cell
as the cell voltage.
Presto-chango, released energy {eV x (# of electrons)}, which in most exothermic reactions would be lost as heat, now is available outside of the cell in the form of usable DC power. And since nearly all of the released power is dissipated externally, the battery does not (significantly) self heat, meaning the battery is an efficient source of power.
All (practical) batteries run one ionic chemistry reaction at the anode that releases electrons (to its terminal) and a 2nd complementary ionic chemistry reaction at the cathode that accepts electrons (from its terminal). Both of these chemical reactions add to the battery voltage and both release power when the battery is discharging. The requirement that (positive) current must be able to flow through the battery internally (from anode to the cathode) means that the anode chemical reactions must release positive ions into solution and/or remove negative ions from solution. And similarly the cathode reactions must remove positive ions from solution and/or release negative ions into solution.
For example at the anode electrons will be released if an active material, like zinc, is used. At the zinc-electrolyte interface zinc's atoms thermodynamically 'prefer' to break up into positive ions (zn2+) able to dissolve into a (polar) electrolyte while leaving the released electrons on the terminal. At the cathode electrons will be absorbed if an electrolyte is used that contains positive metal ions, like copper ions (cu2+), that thermodynamically 'prefer' to snatch up any free electrons and convert to neutral metal atoms as they come come out of solution (either plating out on the terminal or precipitating out). This is the basis of the simple zinc-copper battery (Daniell cell) where positive ions conduct the current at both terminals (into and out of solution), and at the separator current is handled by a reverse flow of negative sulfate ions (SO4 2-) provided by the electrolytes.
More complex reactions that create negative ions, like OH-, at the cathode and destroy them at the anode also work because they provide for negative ionic current flow from cathode to anode. This is the basis of the silver oxide and zinc air batteries. In a lithium ion battery current is carried only by the positive ion Li+, which 'comes out of' the anode material and 'goes into' the cathode material, driven by what Wikipedia calls 'phase changes' in the electrode materials.
When an external load, like a resistor, is added between the terminals, electrons can flow from the anode terminal to the cathode terminal. This allows the reactions at both terminals to proceed because the electrons freed at the anode reaction are bled off and the electrons needed by the cathode reaction are provided. A critical aspect of a battery is that with no load connected the chemical reactions at both terminals quickly shut down due to the build up of space charge layers at both terminals that generate canceling drift current flows. The battery is self-regulating, no load (no current) means no power is released.
What I learned
I figured
out some interesting stuff about batteries, but finding my contributions
in this now wildly overgrown mess of an essay is hard, so (mostly from
memory) here's yet another intro/summary/overview of good battery stuff:
Batteries (at least concepturally) are a combination of a metal and a non-metal. The metal reacts with the electrolyte at the anode releasing electrons (it is oxidized), and the non-metal reacts with the electrolyte at the cathode accepting electrons (it is reduced). Ions generated by the two reactions conduct the current between the two electrodes by moving (drifting under the influence of a weak electric field) through the electrolyte, which is can be, and often is, two electrolytes (in paste or liquid form) separated by an conductive separator. The components of the ionic current can differ throughout the battery, being some combination of positive ions drifting from toward cathode and negative ions drifting toward the anode, which at every distance between the anode and cathode must sum to the external battery current.
The cathode tends to be more complicated than the anode because non-metals tend to be poor conductors. It's common to see the active non-metal cathode material mixed with a conductor, for example, manganese dioxide is mixed with carbon in alkaline batteries. Cathode reactions are often like those in electrochemistry cells where metal oxides are reduced to pure metals, for example silver oxide reduced to pure silver in a silver oxide battery.
The electrode reactions, which generate the battery voltage, occur in fantastically thin layers (a nm or so thick, just a few atoms thick) adjacent to the surfaces of the anode and cathode materials. In all practical batteries the anode and cathode reactions both contribute to the battery voltage, but the oxidizing anode reaction generally the stronger. For example, in zinc family of batteries zinc oxidation at the anode contributes 75%-90% of the cell voltage.
The reactions at both electrodes produce charge separations, so there is a voltage and E field across the thin reaction layers. These voltages are the standard oxidizing (anode) or reduction (cathode) potential of the reactions (adjusted for the concentrations). In my atomic level picture the reacting atoms picking up and dropping electrons do so while part of the electrodes or in physical contact with them, and this is a critical aspect of why batteries work. For example, a zinc ion (zn2+) breaking away from the zinc metal lattice as it goes into solution leaves behind its two electrons in the zinc sea of electrons, giving the zinc electrode a (net) negative charge, which can be bled off externally.
My circuit model of a battery aligns nicely with the battery physically. The model is two batteries in series, one for each electrode reaction separated by a low ohmic resistance (ESR) of the electrolyte. (Arguments I have seen in chemistry lectures that the potential of the electrolyte is much more positive than either terminal is either just plain wrong, or is a red herring.) Both batteries contribute to the cell voltage and power each according to the standard reduction potential of its reactants. A small fraction of the generated voltage drops across the ohmic resistance of the electrolyte and is used to drive (drift) ions through the electrolyte(s) and separator.
The internal space charge batteries at both electrodes are always present. When the cell is open circuit, there is equilibrium and no net current flow, since the local E fields balance the diffusion potentials (in a nernst sense). The space charge local batteries produce the cell open circuit voltage.
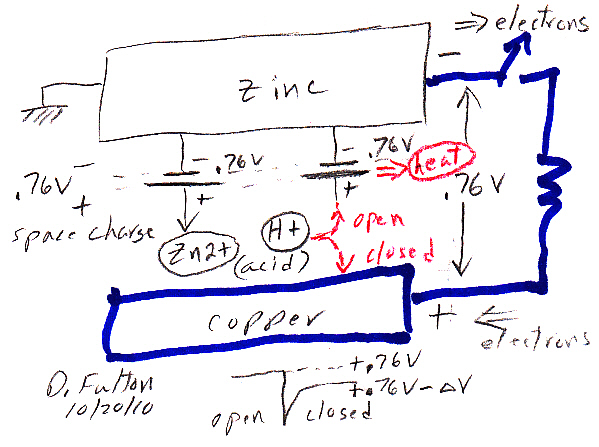
Volta cell (w/acid electrolyte) showing self discharge
H+ anode current
When a load is connected, I have drawn a circuit sketch showing how the external voltage will be just slightly less then the internal space battery voltages, so it is the preferred path for the current. Thus the electrons flow externally from the anode to the cathode dropping through the combined reduction/oxidation potential of the two reactions doing work, rather than internally through the batteries, where only heat would be produced.
Batteries need to be designed so that the reaction products generated don't gum up the works. In the early days when H+ ions in an acid electrolyte carried current, they got reduced to neutral hydrogen at the cathode and slowly obstructed by covering it with small bubble of hydrogen gas. This problem was called polarization. Various anti-polarization measures are now used when H+ is a current carrying ion.
Often the electrolytes have to accommodate the 'waste' product, for example in the zinc/copper daniell (telegraph) battery the zinc anode slowly dissolves into its electrolyte filling it with zn2+ ions. In other cases the electrolyte provides the source of the reactant, for example the daniell battery the source of the cu2+ ions that slowly plate out onto the copper cathode is its electrolyte which starts as a saturated copper sulfate solution.
Many of the common small household batteries (flashlight, clocks, watches, hearing aids) are built around zinc with it's anode reaction provides a large fraction of the voltage. Even batteries without zinc in the name are members of the zinc family: silver oxide, mercury oxide, alkaline, as well as zinc air. The zinc is related to mercury and both have relatively weak bonds between their atoms (low lattice energy). As a result it is energetic favorable (Eo=0.76V) for zinc to dissolve in polar electrolytes. If OH- ions are available from a basic electrolyte, it is even more favorable (Eo=1.25V) for zinc to oxidize to zinc oxide stealing an oxygen from an OH- ion with the remaining H+ joining an OH- ion to make water. (A dissolving solid increases system entropy (S), which because [G = H - TS] helps to drive gibbs free energy (G) negative so a reaction will go.)
Zinc's two battery reactionsIn lithium ion batteries lithium is the anode. Lithium is a group I element, so it really wants to dump its electron. The oxidation voltage of lithium is about as high as any element [Li => Li+ + e- Eo=3.04V], so with such a high anode battery voltage, lithium batteries nearly all have cell voltages in excess of 3V.
Zinc exothermally oxidizes (releases electrons) in both an acidic and basic electrolyte. Early zinc batteries used acidic electrolytes, but most modern zinc batteries use basic electrolytes.a) In an acidic electrolyte metallic zinc dissolves to zn2+ ions
zn => zn2+ + 2e- Eo= +0.76V
b)In a basic electrolyte OH- ions from the electrolyte oxidize zinc to zinc oxide
2Zn + 4OH => 2ZnO + 2H2O + 4e Eo = +1.25V
Some batteries (like silver oxide, mercury oxide and zinc air) have a certain elegance in their equations. The reactions at the two electrodes are partially complementary. All the current is carried by OH- ions that are generated at the cathode and consumed at the anode. To make OH- ions at the cathode water is used (two H for each O) and a source of extra O is needed. In the zinc air battery the extra oxygen comes in through holes in the can, which surprisingly when reducing electons are provided exothermally reacts with the water to make OH- ions. In silver (and mecury) oxide batteries, which are sealed, the extra oygen comes from a metal oxide (like AgO) at the cathode that is reduced to pure metal releasing the oxygen to react with water in the cell to make OH- ions.
At the anode the OH- ions oxides the metallic zinc to zinc oxide (ZnO) with the remaining O and H forming water. Thus equal quantities of OH- and water are generated and consumed in the battery. Silver oxide and zinc air have very flat discharge curves. I don't know that the complementary equations affect this, but it is suggestive. But even these batteries have a disposal problem with zinc oxidizing to zinc oxide at the anode and silver oxide reducing metallic silver at the cathode.
An old (1859) and usual battery is the (car) lead acid battery. It's very unusual in that it has only one electrolyte (so no separator), it's rechargeable, it's not part of any battery family, and has a high cell voltage too (2V). This battery is also unusual in that most (80% or 1.6V) of the battery power comes from the reduction reaction at the cathode. The two plates of a (charged) lead-acid battery are lead (Pb) and lead oxide (PbO2). Both want to react with its sulfuric acid electrolyte (HSO4- + H+). At the anode in an oxidizing reaction the lead reacts with HSO4- ion forming lead sulfate (PbSO4) and spitting back the H+ ion. At the cathode in a reduction reaction lead oxide pulls in HSO4- and 3H+ ions from solution converting to lead sulfate (PbSO4) and spitting out water.
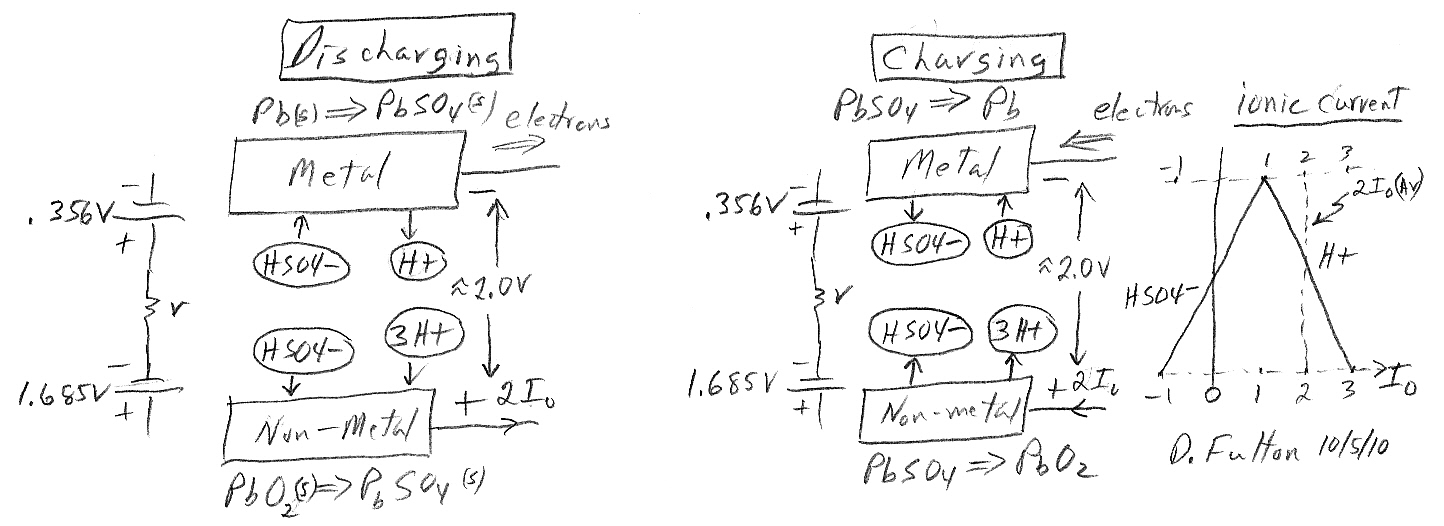
When fully discharged, the anode and cathode plates become mostly the the same (lead sulfate, PbSO4) and electrolyte (stripped of HSO4- and H+) becomes mostly water. When the battery is charging, incoming electrons reduce half the plates from lead sulfate to pure lead (similar to an electrochemistry reduction of lead?) and departing electrons oxidize the other half of the plates from lead sulfate to lead oxide. The sulfate ions and (net) hydrogen released at the terminal reactions reconstitute the sulfuric acid electrolyte.
The role of battery separators is interesting. In general they are thin layers of material used to separate two electrolytes while at the same time allowing ions to pass through. A separator divides the battery into two chambers allowing the chemistry of each half to be optimized, each electrode to have its own electrolyte. A few batteries, like lithium ion and lead acid, have separators, but they don't affect the chemistry. They are just there to provide a physical barrier between the anode to cathode to prevent shorts. In some liquid batteries, like the old Daniell cell used in the telegraph, the electrolyte separation function is provided by liquids with different densities that don't mix.
One super high power, rechargeable battery (sodium-sulfur battery) has reached commercial production and others are at the lab stage. Their purpose is to smooth power from wind and solar. High power batteries usually operate with liquid, molten materials at high temperature and have a simple structure. The cathodes need to be highly conductive. This can be achieved by use of a molten salt, like NaS in the sodium sulfur battery, or a (molten) conducting quasi-metal (a metalloid) acting as a non-metal by accepting electrons, which is the trick used in the liquid metal battery to make a high current cathode.
An interesting new battery, now only at the lab stage, is the liquid metal battery invented recently by MIT Prof Don Sadoway. (Hearing Prof Sadoway speak excited my interest to learn about batteries.) Harking back to the zinc-carbon telegraph battery, the liquid metal battery is a self assembling liquid battery, and because it is (loosely) derived from aluminum smelting cells it is scalable to very high currents and power. Unlike the sodium sulfur battery the liquid metal battery has a simple chemistry with one electrolyte and no separator. It's just three (hot) liquid layers the self separate because they doesn't mix and have different densities (something like oil and water). Hot molten magnesium is the anode terminal and hot molten metalloid antimony (acting like a non-metal) is the cathode terminal. As the battery discharges, both terminal metals ionize [Mg => Mg2+ + 2e-, Sb + 3e- => Sb3-] into a salt electrolyte that floats between them forming magnesium antimonide. Charging reverses the process, separating the magnesium antimonide back into ions that separate and rejoin the liquid metal terminals.
30 Mw of sodium-sulfur batteries have been installed in a solar field in Japan to smooth its output over 24 hours. This battery is composed of two molten liquids (Na and S) with a solid (ceramic) separator between. The two materials combine (Na+ passes through the separator) to make NaS salt in the S cathode chamber. In the liquid metal battery the role of the separator is replaced by liquids whose different densities keeps them separate. In this battery the magnesium anode ionizes and mixes with the metalloid (non-metal) antimony ions from the cathode to make a salt (MgSb) that floats in between the anode and cathode layers. In both batteries the charging process breaks apart the salt and drives their ions back to join the anode and cathode as neutral atoms.
Cathodic protection of ships is done by making a zinc/steel/seawater electrolyte battery that is shorted. Some zinc (anode) is hard bolted (shorted) to the ship's steel (cathode), and the seawater between them is the electrolyte. With the battery terminals shorted the battery voltage drops across the high ohmic impedance of the seawater electrolyte causing a small (ua to ma) positive current to continuous flow into the steel (see my sketch). The inward (positive) current flow prevents the steel from dissolving (corroding) into Fe2+ ions, because any Fe2+ ions created replate back onto the steel. For larger structures where more current (amps) are needed a power supply is inserted in series with the short to raise the voltage, but the principle is the same.
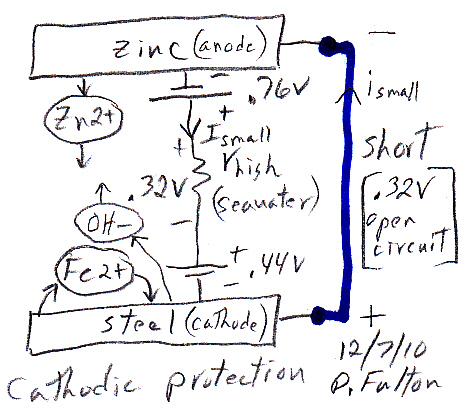
Drawn this way it can be seen that (passive) cathodic
protection forms
a (distributed) battery whose anode is shorted (blue)
to the cathode,
because zinc is bolted to the steel.
(The high impedance of the seawater 'electrolyte'
limits the current.)
Intro
Leaning about
battery electrochemistry has not been easy. I approach batteries as an
EE. I want to draw a battery circuit model, a model tied to the electrochemistry,
to understand the reactions in a battery that generate the power, where
they are located, the atomic picture at the electrode interfaces, how the
ions move, and how to do some calculations. I wanted to understand the
key 'trick' to batteries --- Why does the energy released by the exothermic
chemical reactions comes out as electrical power instead of heat?
Unfortunately electrochemists have their own way of describing batteries,
they don't seem to address the big picture (key trick), they rarely use
circuit models, they use a specialized terminology (which must be learned),
and they tend to gloss over a lot of details I would like explained, and
I got faked out in the beginning by some introductory lecture material
(on the potential of electrolytes) that is both irrelevant and misleading.
Invention
Batteries are not
natural things. They are an invention. A great invention by an Italian
professor, Volta, in 1800. (MIT electrochemistry professor Sadoway jokes
Volta showed that professors aren't useless!) Volta didn't just make the
first battery cell, he came up with a practical way of making a stack (as
big as you wanted) of battery cells such that the cell voltages added.
A big Volta battery 'pile' could deliver substantial voltage and power,
and with this new tool electrochemistry was off and running. Running DC
current through a conducting liquid forces positive and negative ions in
the liquid to opposite electrodes where they are neutralized and (with
good design) can be collected. This technique allows gases and metals of
high purity to be separated out from compounds and natural ores, and very
soon after Volta demonstrated his pile water was separated into oxygen
and hydrogen and several new elements were identified.
Battery as a reverse electrochemical cell
Some perspective
on batteries can be obtained by considering electrochemistry cells. Electrochemistry
cells and batteries are very similar: both have an anode and cathode separated
by electrolyte(s). Both oxidize at the anode and reduce at the cathode,
and current flows through the electrolyte from anode to cathode. The difference
between them is in the polarity of the voltage. In a battery current flows
out of the positive terminal while in an electrochemistry cell current
flows in. In an electrochemistry cell the electrode reactions are not spontaneous
they must be forced (energy supplied), whereas in a battery with a load
connected the electrode reactions are spontaneous.
What is interesting about an electrochemistry cell is that separates and purifies many materials. Prof Sadoway jokes you run current through dirt and you get metals! Electrolytes used in electrochemistry cells (& batteries) are highly polar liquids (salts, acids, bases) that ionize materials, so when a little voltage is applied positive ions in solution are driven one way and negative ions the other. At the cathode positive metal ions join up with incoming electrons and are reduced (neutralized) to pure metal. This must have fascinated early researchers. When connected to a Volta pile for power, it was a chemical tool like they had never seen before, and they used it to discover a lot of new elements in the early 1800's.
Conceptually the way you convert an electrochemistry cell into a battery is by changing the materials so that oxidation and reduction reactions at the electrodes change from endothermic to exothermic, from needing to be driven to thermodynamically 'wanting' to dissolve at the anode and 'wanting' to come out of solution at the cathode.
Overview
Inside batteries
are energy releasing (exothermic) chemical reactions, but unlike nearly
all other exothermic chemical reactions the energy here is not released
as heat. Instead the battery's structure and design (amazingly) causes
the energy released by the chemical reactions to be captured in electrical
form and directed outside the cell where it can do work. Even more amazingly
the chemical reactions inside modern batteries are almost entirely (and
automatically) controlled by the current drawn from the battery. No current
and the chemical reactions shut down. Draw current and the chemical reactions
run at just the rate required to supply the energy delivered. Thus batteries
are an efficient source of chemical power available on demand.
Ionic chemistry
Batteries
are based on ionic chemistry. The chemistry of how elements and molecules
take up (or lose) valence electrons to become charged ions, and how ions
are central to molecules and elements being able to dissolve into polar
liquids (electrolytes), like acids, bases, and salts dissolved in water.
In introductory chemistry there is a lot of (glib) talk about atoms gaining and losing electrons. Bonding electrons are shared between atoms and the sharing can be symmetrical or unsymmetrical. In a molecule like O2 the two atoms are the same type, so bonding electrons are shared symmetrically, meaning the orbital volume of a shared electron is centered between the two nuclei, and the probability of finding the electron nearer atom A (vs B) is 50%. If the atoms bonded are different elements, it's likely that the bonding electron's orbital range will be shifted closer to one atom then the other. For example, oxygen pulls bonding electrons in closer than most atoms. But if a shared bonding electron is spending say 70% of its time closer to atom A and 30% of its time closer to atom B, is it correct to say atom A has gained an electron and atom B has lost one. I don't think so, but this seems to be the way a lot of introductary texts are written.
Ionic solidsMolecules or atoms that disassociate into ions in solution, however, have without question gained or lost electron(s), because they have come apart physically into charged fragments. A negative molecular ion, like SO4- (from sulfuric acid), really has gained an electron, and a positive metal ion, like zn2+, really has lost two electrons.
But when I read up on ion chemistry, I found it is pretty accurate to say that the atoms in an ionic solid, like NaCl (salt), really do lose and gain electrons. A salt crystal is a 3d matrix of alternating Na+ and Cl- ions. Quantum mechanical analysis shows the electron orbitals of adjacent atoms in an ionic solid (like salt) only overlap a few percent.The picture is ionized atoms spaced 'edge to edge' with only a little overlap of the orbitals. However, the radius of the ions can be up to a factor of two different from their parent atoms. This is especially true for atoms in group 1 & 2 of the periodic table (like Na and Ca), which when they ionize to positive ions (Na+, Ca2+), shrink in radius as they shed all the electrons in the next shell. For negative ions it is more complicated, from one point of view they swell, from another point of view they stay the same size (see 'Ionic radius' below).
Bare electrons are unable to go into solution directly, so an electron surplus (or deficit) can buildup on the metal electrodes raising (or lowering their potential relative to the electrolyte (solution). Charge balance can only be restored by an external charge flow or by chemical reactions at the metal/electrolyte interface that create or destroy (neutralize) ions in solution.
Bare protions (H+, an atom with no electrons, hence no real size!) can't go into solution either (Is this true?), but you see H+ in ionic equations all the time. This is just shorthand notation. In a water based electrolyte H+ joins up with a water molecule to make H3O+ (hydronium ion).Single electrode reactionsWhoops, what about pure sulfuric acid? It consists of H2SO4 and as a strong acid it nearly 100% self-ionizes. It's often used diluted with water, but is it ever used undiluted? What happens to H+ then??
One
electrode 'battery' --- Zinc dissolving in acid
Zinc plunged
into (concentrated) sulfuric acid dissolves quickly in a strong exothermic
reaction. All the energy is released as heat. The heat can be viewed as
ESR losses in solution as current loops out and back from the zinc. The
outward current is a diffusion current of zn2+ ions as the zinc oxidizes
and leaves electrons on the electrode and sets up a space charge layer
of 0.76V [zn(s) => zn2+ + 2e- +0.76V]. The inward
current is a drift current of acid H+ ions pushed across the space charge
layer by an E field to the electrode where they grab the electrons and
are reduced to H2 gas bubbles. This is not a battery, but it shows zinc
ionizing releases a lot of energy if only this energy can be captured and
not dissipated as heat.
Single zinc electrode reactions
The data is
that zinc reacts spontaneously making heat when placed in sufuric
or hydrocloric acid (YouTube videos) or copper ion solution (college lecture).
As zinc oxidizes and dissolves to zn2+ and diffuse out into solution the
positive ions in solution, H+ or cu2+, are drawn to the zinc electrode
to be reduced
What happens in timesulfuric acid H2SO4 H+ + SO4-
I think what happens when zinc is plunged into the acid (or copper sulfate) is this: Zinc begins to dissolve (oxidize) releasing zn2+ at the surface that diffuse out into the solution and leaving a growing negative charge on the zinc relative to the electrolyte. The E field set up in the electrolyte by the growing negative electrode charge pulls/pushes H+ or cu2+ ions (zn2+ too, but these don't count) from solution to the surface where they are grab electrons and are reduced to neutral hydrogen or copper. The electrode negative voltage grows until steady state is reached, with the diffusing out zn2+ current balanced by the E field drifting in H+ or cu2+ current. The energy released by the zinc dissolving (& any reduction reaction) can be viewed (at least in a circuit model) as being dissipated as heat by ionic current flows in electrolyte ESR (ohmic resistance).
but zinc does not react (apparently) when placed in a solution with zn+ ions (Daniell cell)
-- Spontaneous (single electrode) reaction occurs when zinc metal is placed in a solution of copper ions.
Zn (s) + Cu+2 (aq) => Zn+2 (aq) + Cu(s)
The zinc oxidizes converting into zinc ions (Zn2+) that dissolve in the copper solution. The negative electrode then attracts (positive) copper ions (Cu2+) that plate out on the zinc (or drop to the bottom of the cell as sediment) as copper atoms. The energy released (in this single electrode reaction) is all dissipated as heat. Note the reaction can continue without a space charge layer forming because downward Zn2+ ionic current is cancelled by upward Cu2+ ionic current.
Single
zinc electrode circuit model
I drew up
a circuit diagram (below) for the single zinc electrode in various
solutions showing separate batteries for oxidation and reduction (options)
at the same electrode. The reaction is spontaneous (generating heat) only
if net power is released. The reaction goes for zinc in acid and
copper sulfate, but does not go for zinc in zinc sultfate. Note in the
case of zinc in zinc sulfate the circuit figure shows the net voltage around
its loop is zero (+76V oxidizing is cancelled by -.76V reducing), so no
current flows and no heat is generated.
In the circuit diagram the power released by the zinc dissolving reaction [zn(s) => zn2+ + 2e- Eo = +0.76V] is modelled by power from the (left side) 0.76V zinc reaction battery. The heat generation of the reaction is modelled by circulating current (even though the ions are different!) flowing through ohmic resistance of the electrolyte (modelled by 'r').
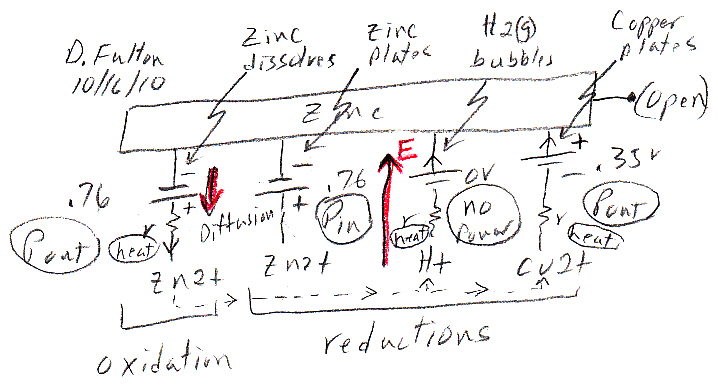
Single zinc electrode circuit sketch for three solutions:
zinc salt (zn2+), acid (H+), or copper salt (cu2+)
Update 10/20/10 --- I now think this schetch
is not quite right.
The 0.76V zinc dissolve space charge extentds across
the 'whole' electrode,
so there should be a 0.76V battery added to the H+
and cu2+ paths.
With H+ and cu2+ exothermic reactions the heat is
lost not from ohmic loses in the ESR of the electrolyte,
but from H+ or cu2+ ions being accelerated by the
E field of the space charge layer,
and then dissipating their kinetic energy as heat
when they slam into the zinc!
Zinc oxidizes (dissolves) to zn2+ creating postive
downward current & releasing energy.
Positive ions in solution (zn2+ or H+ or cu2+) are
attracted to (negative) electrode and are reduced.
Positive upward (drift) current from reducing +ions
'must' balance downward (diffusion) current from oxidizing zn2+ ions.
(zn2+ reduction to zinc absorbs all the released energy)
-- no reaction
(H+ reduction to H2 (gas) requires no energy) -- spontaneous
reaction
(cu2+ reduction to copper releases additional energy)
-- spontaneous reaction
Debye length
There is characteristic
depth for space charge screening called the Debye length. Wikipedia has
an article titled: Debye length. The distance varies inversely to the concentration
and can be meters when thin solar wind gases are considered, but in an
electrolye it is typically in the nm range.
-- Debye length is the scale over which mobile charge carriers (e.g. electrons) screen out electric fields in plasmas and other conductors (including electrolytes). In other words, the Debye length is the distance over which significant charge separation can occur.Formula (for water) is Deybe Length = 0.3 x10^-9 meter/sqrt{concentration in mol/liter}, which is 0.3 nm to 3 nm for concentrations from 1 mol/L to 0.01 mol/L. In other words in water it's about 1 to 10 atomic diameters, or really really thin. I think this distance can be taken as the distance that the space charge layer extends into the electrolyte from the electrode. In other words the local battery in my models (representing the electrode chemical reaction) is physically (probably) an atomic thin layer along the electrolyte, a point which I have never seen made in any reference!
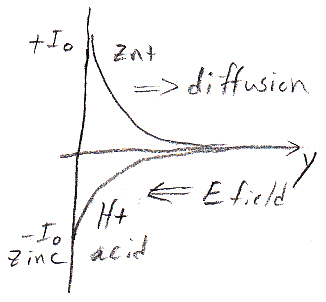
Single zinc electrode in acid
ionic current vs distance from zinc surface
downward zn2+ diffusion current is canceled by upward
drift (E field driven) H+ current
I suspect the length constant here is the Debye length,
which in water would be around a nm or so
OK I think this set of reactions is easily explained. Whether the reaction (zinc dissolving) is spontaneous must depends the sum of the (zinc) oxidization voltage and the positive ion reduction. As shown below zinc with zn2+ ions has a sum of exactly zero volts, it doesn't go (this explains why Daniell puts zinc in zinc sulfate). But with H+ there is +.76V and with cu+ there is +1.10V so both reactions are spontaneous.
oxidation zn => zn2+ + 2e +0.76V
reduction
zn2+ + 2e => zn(s)
-0.76V
H+ + e- => H2(g)
0.0 V
cu2+ + 2e- => cu(s)
+0.35V
Zinc
in copper sulfate solution
In a chemistry
book there was a question about what happens if a piece of zinc is placed
into a copper sulfate solution (1 mol/L) and just left until equilibrium.
Ans: All the zinc (gradually) dissolves going into solution as zinc ions
(1 mol/L) to be replaced at the metal by copper ions plating out from the
solution. No other details were given.
Like in the zinc in acid case no space charge builds up to stop the zinc from dissolving, because the solution contains a lot of positive ions (cu2+) that can react (reduce) at the metal surface and provide a back current. This is a one electrode system, so the zn2+ current from metal to solution (as the zinc oxidizes and releases electrons) must be matched exactly (says Faraday) by the back cu2+ current from solution to metal (as the copper ions grab free electrons and get reduced to neutral copper atoms). Here's my sketch showing the current flow:
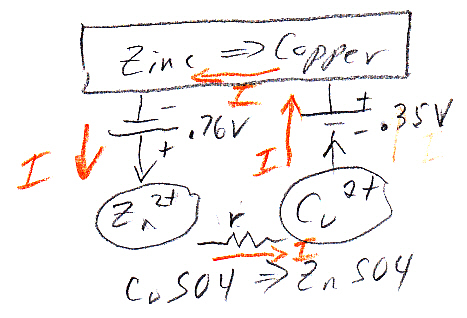
my model of current flows of a 'one electrode' zinc-copper
system
(zinc metal dissolves into copper sulfate and copper
ions plate out)
Notice the current flows flows in a loop (really lots of little loops). The model shows the two batteries of the chemical reactions are both putting out power [P = I x (.76V zinc oxidize + .35V copper reduce)], as they would in a copper-zinc battery, but where does the power go? The only place I can see it going is as heat into the solution, which in the model shows up as power lost in the electrolyte resistance (r). (I am not entirely satisfied with this explanation, that heat is dissipated in ohmic resistance of the solution. Is there (perhaps) some heat absorbed in a phase change?)
Ion level picture of battery chemical reactions
A central
mystery of how batteries work is this --- How does the (presumably)
very short range voltage from E fields from atoms involved in the battery
chemical reactions get (efficiently) transferred (or coupled) to the battery
terminals? To understnad this, to 'really'' understand how batteries work,
I want a picture that shows at the ion level how this happens. No reference
I have found addresses this, but after learning some ion chemistry and
thinking it through, I have come up with the picture belows, one for the
cathode and one for the anode.
The closest some references come to addressing the voltage issue is stating that charge separations, with their attendent E fields and voltages, set up along the surfaces of the terminals to limit the rate of the chemical reactions. The voltages across these charge separations correspond to the local 'batteries' in circuit models, and the sum of the voltages across the two charge separations 'batteries' is what appears at the terminals. This is OK as far as it goes, but I would like a deeper explanation, at the atomic level. What do you see if you sit on an atom/ion involved in the chemical reactions?I'll use as my example, probably the simplest battery, the zinc-copper daniell cell. And I think it's valid to employ a standard 'trick' of circuit analysis: superposition. Superposition says circuits can be analyzed by 'turning on' voltage sources one at a time (with the others voltage sources shorted), and then summing the results. The battery analog is to treat the two chemical terminal reactions separately.
In the daniell cell positive metal ions (cu2+) come out of solution at the cathode and plate the terminal generating a voltage of 0.35V. It seems reasonable to argue that in some sense the (negative) electrons in the (external) load are 'attracted to' the positive metal ions at the cathode. But this doesn't really help the picture because it doesn't address how the short range E field voltage around the ions gets transferred to the terminal. It seems to me it can't be that the incoming cathode electrons 'jump' off the cathode to the incoming cu2+ ions neutralizing them, because then the electrons would see all the ion voltage as they fall into the atom neutralizing it. What does seem to make sense is that incoming positive metal ions (cu2+), driven by a weak E field in the electrolyte, first make contact with the cathode terminal, transferring their positive voltage to the terminal, and then they accept two electrons becoming a neutral ion.
Cathode picture
If we think about
copper metal how this happens becomes clearer. Good conducting metals like
copper are described as having a 'sea' of electrons. In other words one
(or more) of the valence electrons of many (all?) copper are not attached
to particular atoms. They float around and are shared by many atoms. This
is why a small (external) voltage applied to copper causes a large current
to flow, the sea of electrons move under the applied E field. But this
must mean that immobile atoms of metallic copper must look like a matrix
of positive ions with their positive charge on average balanced out by
the sea of mobile electrons.
So a simple picture emerges. The cu2+ ions coming in from solution are probably just like the positive ions that make up the metallic copper matrix and are able to just accrete to (or fit into) the immobile matrix. There is really no need to specifically neutralize each incoming positive ion! When copper ions come in from solution and join the matrix (without adding electrons to the electron sea), it makes an unbalance in charge on the whole copper cathode terminal. Ther is (slightly) more positive charge than negative charge. This positive charge unbalance causes the cathode terminal voltage to rise (relative the anode) and the positive cathode terminal then 'pulls' (attracts) load electrons toward it. As incoming electrons from the load add to the sea of electrons, more positive ions come in from solution to keep the cathode voltage at a stable positive voltage (0.35V). I think this picture of the cathode hangs together pretty well.
There is potential energy associate with the crystal structures of solids. Think about the ice-water transition. To melt ice heat must be added and temperature does not rise (stays at 0C). Hence atoms in a solid crystal structure are at lower energy than when in a liquid. When a liquid transitions to a solid energy must be released as the atoms settle into a lower potential energy environment. So in the case of copper growing its matrix, it could be argued that the energy released by the cathode reaction comes from the growing matrix. (I'm not sure this arugment though makes any sense, since an ion coming out of solution might be very different from a simple phase transition.)Anode picture
When I check the resistivities of zinc and copper I find that zinc is also a good conductor, but not as good as copper. The resistivity of zinc is x3.5 higher than copper, but since zinc is a pretty good metallic conductor, it's likely that the 'sea of electrons' picture applies to it too. However, it's likely that its electron sea is a little sticker, a little less mobile than the sea in copper. This is a guess, but it might be that in the case of copper thermal forces keep the copper electron sea really unstuck from atoms, whereas in zinc they may be loosely bound. (complete guess).
Battery definitions
A (basic)
battery consists of two electrodes with a conductive ionic solution (or
paste) between them with materials chosen so that an open circuit voltage
appears between the electrodes and which will supply current (& power)
when an impedance (load) is place across the cell. The anode (negative
terminal) is made from a metal that reacts spontaneously (exothermally,
releasing power) with ions in the electrolyte if electrons are drawn off
the electrode. The cathode (positive terminal) is made from a non-metal
(or a real metal like copper acting like a non-metal) that reacts spontaneously
(exothermally, releasing power) with ions in the electrolyte if electrons
are input to the electrode.
While conceptually a battery is formed if the two electrode reactions produce net power, i.e. the power out from one electrode reaction exceeds the power consumedby the other electrode reaction, in all practical batteries (I suspect) both electrode reactions contribute to the power out of the battery.
Anode
* Anode electrode
is typically a metal, and its electrolyte interface reaction is
oxidizing
releasing electrons to the terminal. In discharge positive ions are released
to, and/or negative ions accepted from, the electrolyte. Anode metals often
dissolve
into the electrolyte. Electron flow is out of the anode, so it is
the negative terminal of the battery.
Cathode
* Cathode electrode
is typically a non-metal (or effectively a non-metal), and its electrolyte
interface reaction is reducing accepting electrons coming into the
terminal. In discharge positive ions are accepted from, and/or negative
ions released to, the electrolyte. Positive metal ions in solution often
plate onto the cathode, and sometimes at the cathode hydrogen gas is released
as acid H+ ions get reduced. Electron flow is into the cathode so
it is the positive terminal of the battery.
Circuit model
* The battery circuit
model is two batteries in series, one for each electrode/electrolyte interface
reaction, and between them an (ohmic) resistor modeling the resistance
of electrolyte and separator. In all practical batteries (I suspect) both
electrode batteries point the
same way, meaning both electrode
reactions (are spontaneous and) contribute to the power of the battery.
In open circuit state the potential of the electrolyte is uniform, i.e.
there is no potential drop across the electrolyte, and the electrolyte
potential is intermediate between the positive and negative terminal voltages.
During discharge ion motion is (I suspect) largely driven by E field (the
voltage drop across the electrolyte resistance) with perhaps diffusion
important near the interfaces and away from the main ionic path.
Standard reduction potential (Eo)
* The voltages
of the electrode batteries in the circuit model can be figured from the
'standard reduction potential' (Eo) table (corrected for non-standard conditions),
which is a tabulated list of measured electrode voltages (in volts)
for various electrode/electrolyte reduction reactions against a hydrogen
standard cell. For oxidizing reactions (at anode with release of electrons)
the voltage is just the negative of the reduction potential. Standard reduction
potentials range from -3V (lithium) to +3V (fluorine) with hydrogen (reduction
and oxidation) by definition in the middle at 0V. Thus theoretically the
maximum voltage of any cell is 6V equal to the sum of a +3V lithium anode
battery and a +3V fluorine cathode battery. (some practical lithium cells
are 3V)
Electrolyte
Electrolytes
are ionic solutions. This includes acids (lots of H+), bases (lots of OH-),
molten salts, and (what ever class zinc and copper sulfate are in??). Electrolyte
ions nearly always are play a role in the battery chemistry, and the ions
in solution by their motion carry the current across the cell. A battery
can have one electrolyte, like a car lead acid battery, or more commonly
two electrolytes with a separator in between. With a separator there is
much more freedom to pick and optimize the reactions at each electrode.
Electrolyte acids like sulfuric acid (H2SO4) have very polar molecules. These (liquid) materials strongly ionize not because the molecules 'fall apart', but because the strong dipole forces between them allows the negative end of the molecule to occasionally rip away an H+ from a neighbor. An autoprotolysis equilibrium constant (or 'acid disassociation' constant or 'self-ionization' constant) of about 10^-4 for H2SO4 indicates that in equilibrium about 1% of H2SO4 molecules are positive H+ ions (really H3SO4+) and 1% HSO4- ions. (Compare this to water, also quite polar, where the autoprotolysis equilibrium constant is 10^-14 indicating that only one in ten million water molecules are ionized.)
Separators & salt bridges
Separators
and salt bridges are used to electrically couple two electrolytes together
(via ion flow). A salt bridge is a high impedance device (1k or so, long
narrow tube filled with a salt electrolyte) used only in the lab to measure
'open circuit' cell voltages. Separators are (I think) sort of a low impedance
version of the salt bridge used in real batteries. A separator is usually
a thin layer (of various materials) that allows some ionic diffusion through
it. (whether its ionicly selective I do not know)
(Haven't read much about separators, but I suspect they impose a resistance to some ions and perhaps block others. I wouldn't be surprised if like many membranes they develop a nernst voltage across them. Is this a 3rd battery in the model? or is it just a resistor. I have seen no mention of a separator potential, so I am assuming a separator can be modeled by an added resistor in the electrolyte path, which means extra heat lost in the battery during high discharge.
How
do batteries make chemical power available to do work?
At the atomic
level energy is released when (valence) electrons give up potential energy
as they move to a lower voltage nearer the nucleus. The electrons see a
real
voltage change as the move through the E field of the atom. eV (electron
volt) is a unit of energy, so the (net) voltage change of the electrons
is a direct measure of the energy released by (or input to) a chemical
reaction.
While perhaps oversimplified, the equations of redox reactions can be separated into two steps: an oxidation step, where electrons are released, and a reduction step, where those electrons are gathered in. The cleverness of the battery invention is that those two chemical reaction 'steps' are made to occur in separate locations: oxidation reactions at the anode/electrolyte interface and reduction reactions at the cathode/electrolyte interface. The electrons released by oxidation are collected by the anode electrode, drop through a voltage (in an external load) doing work, then arrive (spent of energy) at the cathode electrode to support reduction.
The beauty part of a battery, or really two half-cells, is that the oxidation and reduction voltages are exposed and directly measurable (with a volt meter). Physically the half-cell voltages are generated by a space charge separation layer that develops (at each electrode) to bring diffusion and electrical drift currents into balance. From the measured half-cell voltages circuit models can then be drawn showing the details of where in the battery the energy is coming from, and importantly, how the external load path is preferred by the electrons over the internal (heat generating) path.
Heat or work?
I spent a
long time thinking about how just adding a 2nd electrode to a zinc/acid
system, like Volta did, can stop a runaway zinc dissolve in acid heat reaction.
In fact I don't think just adding an extra electrode without hooking it
up, i.e. open circuit, makes any difference. How could it? If zinc is exothermally
dissolving in acid, what difference can it make if a piece of copper, which
does not react with acid, is inserted too? But this misses the point. The
real issue is can an extra electrode kill the zinc dissolve heat reaction
if a load is connected, and I think the answer is, yes it can.
From a slightly different viewpoint: How can running some current between the zinc and a copper electrodes shut down the 'self discharge' internal exothermic reaction of zinc dissolving in acid, where all the energy released goes into heat? Or how can adding an extra electrode and load produce a real battery where all (or nearly all) the chemical energy is available externally to do work?
If I understood this, I figured I would have a good handle on understanding how batteries really work. I found almost no discussion of this online, but thinking through the physics/chemistry of this case, I came up with the following sketch.
Volta cell (w/acid electrolyte) showing self discharge
H+ anode current
H+ acid ions are at a potential 0.76V above the (negative)
zinc electrode (here shown connected to ground).
When switch closes, a source of electrons for H+ reduction
also becomes available at the copper cathode,
but at a slightly lower voltage than 0.76V.
Thus the H+ acid ions redirect their flow toward the
lower voltage copper electrode
completing a (positive) ionic current flow through
the battery.
The key as to whether the chemical energy released by zinc oxidizing and ionizing into solution is lost as heat or is made available externally (at battery terminals) to do work depends on the path of the acid H+ ion flow, i.e. how the H+ ions complete the circuit. I show the H+ path options in red in my sketch above.
Upward path ('open') --- Acid H+ ion flow across space charge layer to the zinc anode dissipates the zinc oxidizing chemical energy as heat.Space charge layer formsDownward path ('closed') --- Acid H+ ion flow to the copper cathode makes the zinc oxidizing chemical energy available (externally) for work.
In the sketch the space charge layer is represented by two 0.76V battery spaced out along the zinc electrode. As the zinc (slowly) dissolves, the zinc oxidize reaction [zn(s) => zn2+ + 2e- Eo= +0.76V] delivers a steam of zn2+ ions into solution (near the surface) where they diffuse away. (They can't be ejected into the acid with too much velocity, because all the zn2+ kinetic energy is going to be lost as heat.)
a) Switch open
The
added electrode and load are in blue, but initially the switch is open,
so no current flows externally. With the switch open the H+ ions of the
acid, which are attracted to the negative zinc terminal, have only one
path to the zinc and that is across the space charge layer, so the exothermal
zinc dissolving reaction continues as though the copper electrode was not
there. The zinc must dissolve much slowly here than you see in YouTube
videos, presumably because the acid concentration is probably much lower,
and it's not liquid, but liquid dispersed in cardboard.
The left battery in the sketch has a (positive) current of zn2+ ions flowing out of the positive battery terminal. This battery is delivering energy, and the energy coming out of this battery represents the energy of the chemical (zinc oxidizing) reaction.
The right battery has a (positive) canceling current consisting of acid H+ ions flowing to the zinc, where they are pick up electrons left by the ionizing zinc atoms and get reduced to H2 (hydrogen) gas. The polarity of the right battery is the same as the left battery, but here the (positive) H+ ionic current is into the positive battery terminal. The H+ current flow is delivering power to the right battery. What does this mean? It means the right battery 'power in' represents the energy being dissipated by the reaction as heat. Charge neutrality requires the (average) H+ current to the zinc be exactly equal to the zn2+ current from the zinc, so with the switch open the heat dissipated in the right battery is exactly equal to the energy flowing out of the left battery (modeling the release of chemical energy from the zinc oxidation).
A simple physical atomic picture of the heat generation is this: Slow moving H+ ions in the acid fall into the E field of the space charge layer. They get accelerated by the space charge E field (below I calculate it to be 3,000 m/sec), which does work on them, then they slam into the zinc metal dissipating all the kinetic energy they acquired as heat. In other words the chemical zinc dissolve energy first gets stored in the space charge layer E field, second, the E field gives up some of its energy accelerating positive H+ ions across the space charge layer, and three, the work done on the H+ ions by the E field gets dissipated as heat when they slam into the zinc metal.
Note, this represents a change in my thinking. At first I though the exothermic heat of the zinc/acid dissolve reaction could be modeled as heat loses in the ESR of the electrolyte as ionic currents circulated, but thinking it through I now am pretty sure that the heat is due to the H+ ions picking up energy as they cross the space charge layer E field which they dump as heat when they decelerate. From a circuit modeling perspective this means the heat loss should be represented not as heat in an ohmic resistor (ESR), but as current (H+ ion flow) delivering power to a battery (0.76V of the zinc oxidizing space charge).b) Switch closed
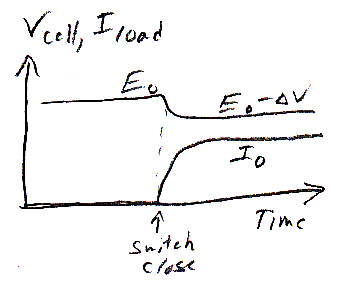
It turns out that the external path is favored. To see why this so (for reference only) I show the zinc electrode grounded, then we can speak of the H+ ions in solution as being 0.76V above ground (potential). When the switch closes, the voltage of the copper electrode will quickly drop, because it will take some time for a high flux of H+ ions (& possibly zn2+) to migrate to the copper electrode. I shows this in the sketch as a step drop in voltage followed by a recovery as the ion flow to the copper increases.
From a circuit point of view a flow of positive ions in solution is a (positive) current. The H+ ionic current has two paths to ground (zinc), one through the 0.76V (space charge) battery and the other through the (ohmic) impedance of the external load. Circuit theory says for the positive ionic current to flow into the positive terminal of the (right side) 0.76V space charge battery (and deliver power to it) the potential must be equal to or in practice (a little) higher than 0.76V. So all the external load need do is to pull the copper potential slightly lower than 0.76V and all the H+ ionic current will divert to the copper path. I show this on the sketch as the copper voltage recovering buy not quite to 0.76V (shown as +0.76V - delta V).
Zn2+ ions at the cathode?
This begs
the question as to whether zn2+ ions contribute to the current flow at
the cathode. In the Volta battery there are two positive ions (zn2+ and
H+) in solution and both are pushed by the (weak) E field in the electrolyte
toward the cathode, and both (in principle) can be reduced at the cathode,
carrying the current into the cathode. I am not a good enough chemist to
really explain this, but I think it's an issue of competing reactions.
The reduction voltage of H+ (0V) is more positive than zn2+ (-0.76V), meaning
H+ is more easily reduced that zn2+. Zn2+ really doesn't want to
accept an electron. And if zinc ions alone had to carry the current between
electrodes, there would be no battery, because to reduce zn2+ ions back
to zinc takes all the energy released when they were created. Hence all
(or virtually all) the current at cathode must be carried by acid H+ ions
being reduced.
Positive current at the zinc/acid interface must all be carried by zn2+ ions and at the acid/copper interface all by H+ ions with a transition between them across the electrolyte. As the battery discharges, zinc must build up in the solution and the acidity level must go down as H+ ions get reduced to hydrogen gas at the cathode.
A key to how batteries work!From a work/heat point of view there is huge difference the two H+ paths. If back ionic current flows internally across the space charge layer all the chemical energy is lost as heat, but if it flows externally it all the chemical energy is available to do useful work! Notice from the figure it takes only a tiny change in electrode voltages to shift the H+ current path from internal to external. And the terminal voltage change needed is generated by the current flow in the load. The external load needs to only slightly lowers the positive copper voltage to divert the H+ current to cathode and outside path.
A key to how batteries work is that connecting load to a battery slightly lowers the voltage between the cathode and anode. This redirects the flow of positive ions toward the cathode (and away from internal feedback paths), and the load provides a flow of electrons at the cathode to reduce ions that arrive. The result is that the current initiated by the chemical reaction (zinc dissolving) at the anode is able to (easily) traverse the electrolyte and the preferred return path is via the external load connected to the battery terminals.For convenience assume the zinc anode (negative terminal) is grounded. Open circuit the cathode sits at a more positive voltage set by the voltage across the internal space charge layer (or layers). Connection of an external load, by pulling the cathode voltage slightly lower than the space charge voltage, redirects positive ions away from the space charge layer toward the cathode and via reduction at the cathode to a return to the zinc anode through the external load. In this way any wasteful heat generating losses in the space charge layer are killed and all the chemical energy is available externally to do work.
Presto, chango, just by letting a little current flow from this simple zinc/copper/acid (Volta) battery its inherent 'self discharge' (heat generation) mechanism is shut down and all the chemical energy is captured for doing work as the H+ ion flow is diverted from the internal space charge layer to the external load path.
Sure the Volta battery has a flaw, a self discharge mechanism due to acid H+ ions allowing the zinc to dissolve even with not load, but when a load is connected the (slightly) lower cathode voltage redirects H+ ion flow and shuts down the internal heat loss mechanism, hence when discharging this is an efficient battery.
Preferred
current path is external
I drew the
sketch below to illustrate how the preferred path for the current, here
generated only by an oxidation reaction at the zinc anode (Volta cell),
prefer to flow back outside the cell, where is does work, rather than flowing
back internally and wasting the released chemical energy as heat.
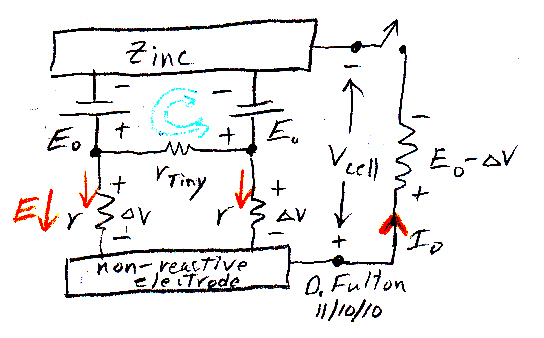
Blue (no load, switch open) --- canceling (small)
diffusion and drift currents across the zinc space charge layer
Red (with load, switch closed) --- Resistor path (R)
is preferred over internal E0 battery path, because resistor
voltage stabilizes as just slightly less than Eo.
In the above sketch the two top Eo batteries represent the very narrow space charge layer that develops right at the surface of the zinc to stop (net) dissolving of the zinc. Blue shows an undefined, but probably small tending to zero, current flow of zinc ions that thermodynamically are forced across the local E field (zinc 'wants' to dissolve). With the switch open this current flow out (from the zinc) is balanced by a current flow in toward the zinc by zn2+ ions in liquid that get caught by the local E field and are pushed back to the zinc electrode. The equilibrium voltage (Eo) that sets up, with canceling diffusion and drift currents, is a classic nernst potential first modeled by Walther Nernst in the early 1900's.
When the switch closes, the outward current from the zinc now has a choice of two paths to return to the zinc to complete the circuit. One path is flow through the Eo battery (i.e. back across the space charge layer), and the second path is through resistors (r +R), electrolyte ohmic resistance plus the external resistance. If the voltage across (r + R) is just slightly less than Eo, then all the current will prefer the external path. And this is what happens in all batteries when a load is applied. The load in a sense sucks the current away from the inside battery path to itself.
The key is this: A positive current can only flow into the positive terminal of an Eo battery if the voltage is equal to (or in practice slightly above) Eo. The resistance path need only have a voltage just slightly lower than Eo to get all the current (and power) of the battery. The slight voltage hang off across the load ('delta V'' in the sketch) drops internally across the ohmic resistance (ESR) of the electrolyte, i.e. it makes a weak E field in the electrolyte between the electrodes, driving positive ions to flow from anode to cathode (& negative ions from cathode to anode) completing the circuit.
Particularly revealing is to compare the circuit model of a single zinc electrode in acid, where all the energy comes out as heat, to the same electrode in a (Volta type) battery with a load, where (nearly) all the energy comes out as electrical work. (something I have never seen in a textbook or online)The space charge/circuit model of a battery makes it clear almost 'by inspection' (as engineers say) as to how it is that the chemical reactions in the battery are controlled by the current drawn from the battery.
How fast are ions accelerated across zinc space charge?
For fun let's
see how fast a positive H+ ion (which doesn't really exist in solution)
would be accelerated crossing a 0.76V zinc space charge layer. (This is
a calculation I have never seen anywhere!) This is basically just a units
problem, finding the velocity (m/sec) for a mass of one unit (proton mass)
to have kinetic energy of 0.76 eV.
E = (1/2) m v^2
v = sqrt{2E/m}
v = sqrt{(2 x 0.76 ev x (1.6 x 10^-19 joule/1 ev))/1.7 x 10^-27 kg}
v = sqrt{(1.4 x 10^8}
v = 1.2 x 10^4 m/sec (for a proton)
= 3 x 10^3 m/sec (for a H3O)
The idealized H+ ion is really an H+ glommed onto water (H3O+), the hydronium ion. The H3O ion speed is about 4.4 times (= sqrt{19} lower than a bare proton. I was wondering if the molecule speed might be up near the speed of light. Well it's not, but it's still pretty fast.
Open questions
There appear
to be some assumed basics, which I don't have the background in chemistry
to understand.
---- One is
no neutral atoms appear to be allowed in solution. Why? If an electrode/anode
metal dissolves I always see it ionizing and the ions go into solution.
---- Why is
it electrons don't go into solution. What blocks them? Obviously for a
battery to work you need to force the electrons to flow externally. Are
they blocked in principle or is this a matter of engineering, are the electrodes
and electrlytes chosen so that there is no atoms they can convert to a
negative ion in solution??
Introduction
After learning
a little chemistry working on photosynthesis and fuels, I've decided to
take a look at batteries. I have no idea how batteries work inside,
but I've been curious about them for a long time.
How do you make a battery?I know nothing about batteries
How do you make a battery? I suspect the heart of the battery is two chemicals that 'want' to join in an exothermic reaction. They have very different electronegativity so as their valence electrons move through the (redox) voltage difference (drop into an energy well of the new compound) a lot of energy (eV) is given off in the form of heat. To make a battery out of these chemicals the valence electrons must have only one path which is through an external circuit, and this allows the energy of the reaction to be captured so it can do useful work.Basic battery
Thinking about it a little more. An electron moves from a metal, where it is loosely held, to a non-metal, where it will be tightly held. This can (at least conceptually) be though of as two reactions in cascade: an oxidation (metal is oxidized, i.e. it loses an electron and become a positive ion) and a reduction (non-metal is reduced, i.e. it gains an electron and becomes negative ion). These will be the reactions at the battery electrodes. The positive metal ion and negative non-metal ion must then diffuse (or be pushed by an E field) through the intermediate electrolyte (and any barrier) to join up forming the new compound (only in the electrolyte?).
The 2nd (and immediate trigger for this essay) was a short MIT video by an MIT professor (Prof Donald Sadoway) who is working to develop a fundamentally new type of battery, a liquid metal battery. He envisions monster cells like in aluminium plants that would handle 500,000 amps. It's use would be grid smoothing of intermittent sources like solar or wind technology. (By coincidence the NY Times had a story two days earlier that wind is driving up the demand for batteries, especially in isolated places with a lot of wind power like Hawaii.)
Sadoway in the video pointed at a color coded periodic chart and said you have 'metals' (most of the chart), which donate electrons and non-metals (right hand corner) like carbon and oxygen, which accept electrons. Elements with different "electronegativity". Connect them together with a conducting salt and you have a battery. Is it really this simple?
Basic battery
I sketched
up what I think is a basic battery. I have seen no write up like this.
All the battery descriptions I see just assume a lot of things I am trying
to make clear here.
The heart of the battery is a metal and non-metal used as the two electrodes (they may be embedded in other materials as matrix, but these are the active electrodes). These two materials if able to mix would react strongly (under what conditions, only as ions??) releasing energy (exothermic) because the non-metal has a nice hole into which the metal semi-free valence electron wants to fall.
But in a battery the two electrodes are physically separated (maybe there is a barrier too) and the only way they can come into contact is by 'dissolving' (is that the right word?) into the electrolyte. And a further requirement for the electrolyte (which may be trivial) is that only ions of the electrode metal and non-metal, and not their neutral atoms, can dissolve in the electrolyte. It appears that the inherent voltage difference in the two materials is now visible at the terminals of the battery in the form of an open circuit voltage. (Is this voltage the absolute electronegativity??) The connection of a load provides a path for electron flow, which in turn converts neutral metal and not-metal atoms at the boundary of the electrodes and the electrolyte into ions (as shown in the sketch). The two electrode ions meet and join in the electrolyte, but not exothemally (no heat release inside the battery). The voltage drop seen by the electrons from the metal transferring to the non-metal occurs external to the battery in the outside load. The metal reaction compound is formed by the positive metal ion and negative non-metal ion joining (probably) ionically. Prior to their joining the electron transfer between the two materials has already taken place as they were transformed into ions at the electrode/electrolyte interface.
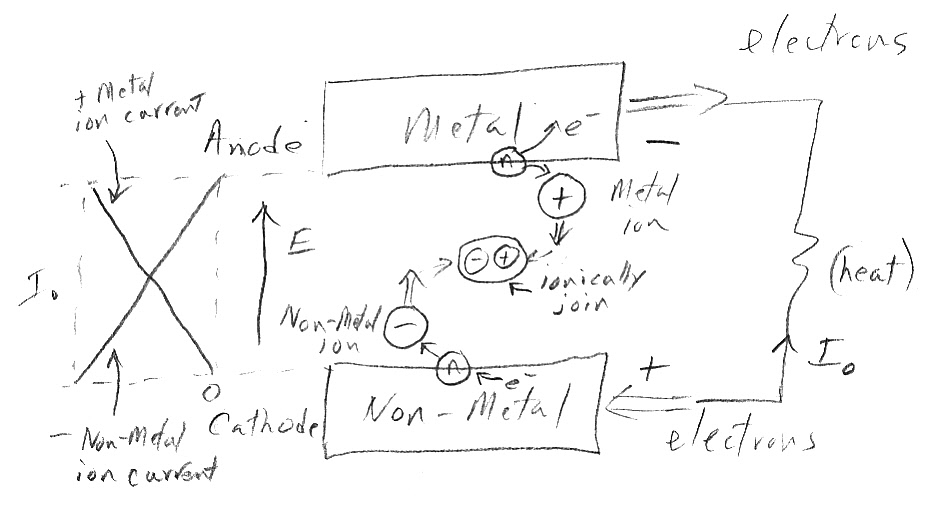
my (2nd) 'basic battery' sketch (9/2010)
metal (top) releases electrons at 'anode'
non-metal (bot) accepts electrons at 'cathode'
(this anode/cathode convention is used by Prof Sadoway)
Looking at the sketch it must be that the ions diffuse away from the electrode (interface) where they are created toward the other electrode. They can't be moving under the influence of the E field, because they are moving opposed to it (confirmed by a comment in Wikipedia that the ions move 'uphill'). A diffusion gradient also makes sense because each electrode/electrolyte interface is pouring out ions. On the left I sketch the two ionic components of current inside the battery. I assumed a linear ion gradient across the battery. This is idealized, I have no idea how close to reality it is. I also have the current inside the battery go from being all metal (positive) ions near the metal electrode to being all non-metal (negative) ions near the non-metal electrode. (Seems like the pos and negative ions must match).
It will be interesing as I read about various batteries whether I can map their chemistry into the sketch above.
Do electrons or protons exist in isolated form
in solution?
The
short answer is probably: no
Electrons and protons are both so tiny that its hard to think of them coexisting with structures that have a nucleus and bound electron(s), i.e. atoms, ions, molecules, all of which have a much larger and well defined radius.
An online chemistry test asked the student to calculate the atomic spacing of gold (solid) and mercury (liquid) giving their densities (kg/m^3) and implied the result were generally applicable. The atomic weight of gold and mercury (adjacent in the periodic chart) are within 4% of each other and the density of mercury was given at about 70% that of gold. I did the calculations and for solid gold I found the atomic spacing is about x2 radii of the various radii listed in Wikipedia. In other words the atoms are packed in (probably) to some degree locked making it a solid.
The moderately lower density of mercury means its atoms are just a little less tightly packed, a 30% density difference only requiring that linear spacing be increased by 10% to 12%. So in liquid mercury with just a 10% or so wider atomic spacing compared to a solid (1.1 x 2 radii), the atoms are able to slide around each other and we have a liquid.
The clear answer from the electrochemisty book (below) about isolated electrons in solution is that they do not exist. All electrons in solution are in the form of negative ions (electrons glomed onto otherwise neutral atoms or molecules). And the equilibrium equations in the book for protons (H+) are in terms of H3O+ = [H2O + H+] implying that the proton too is not isolated, but typically glom onto a water molecule (making H3O+).
======================================================================
References
Search for 'electrons in solution' brought up this book. Looks interesting. Detailed quantum discussion of potentials.
Modern Electrochemistry, Vol2, by Bockris, Reddy, Gamboa-Aldeco (searchable in Amazon)
Book confirms (what I had suspected) that there are no separate (mobile) electrons in liquids, saying in metals electrons are "mobile", but in solution they are not mobile as they are bound to ions. The book says "hydrated electrons" can be created in solution, meaning the electron is "in a state within an ion"
-- hydrogen
electrode potential (unknown prior to 1967).
Calculations put it at 4.6 V +/- 0.2V "Regarded
by most people as the absolute potential of the standard hydrogen
electrode."
-- When a cell is created, the voltage measured is the difference between the potentials of the two electrodes. When electrode potentials are measured (tabulated), what is done is to change the material of one electrode while holding the other electrode constant as a reference. The reference electrode being (at least in principle) the hydrogen electrode. Since the absolute hydrogen electrode potential (now calculated to be 4.6V) is just an offset in the table, in practice it is taken as zero, allowing the measured cell potential to be treated as the potential for the test electrode. (The absolute potential is in refenece to stationary electrons (and protons) in a gas phase, i.e stationary in a vacuum and an infinite distance from particles with which it might interact.)
-- How electrons flow from ions (in solution) to electrodes was figured out (using quantum principles) in 1931 by the first electrochemist, Ronald Gurney. Basically the electrons 'tunnel' through the energy barrier formed between the electrode and the ions adjacent to the electrode.
-- electrons in the metal of a reference electrode are in equilibrium with the protons in solution. At any chemical interface the chemical potentials on the two side come to equilibrium. In a 'charged' interface between a metal and solution the metal and solution electrochemical potentials come into equilibrium. This electrochemical potential has a name: 'Fermi energy of the electron in solution.' (In other words the electrons in the outermost electron levels of ions exchange states states with, and come into equilibrium with, the topmost occupied states or Fermi levels in the metal.)
-- de Broglie's equation for the wavelength of mass
de Broglie wavelength = h/momentum
= (h/mv) x sqrt{1- (v/c)^2} (relativistically)
where
h = 6.6 x 10^-34 joule-sec (planck's constant)
momentum = mass x velocity (non-relativistically)
In solids the de Broglie wavelength must 'fit' atom centers to form standing waves.
-- (Wikipedia) de Broglie wavelength of a thermalized electron in a non-metal at room temperature is about 8 nm. (This is huge. The radius of a typical atoms is about 150 pm.)
-- At room temperature the most important energy in solution is usually the ground vibrational state of the electrons at the ion/metal inerface.
-- Book derives an equation for the electron potential in solution, which for a dilute solution is
ue (electron potential in solution) = -kT ln (fe)
where
fe = "electronic partition function of the
electron in ions in solution"
kT = 25 mv
The form of fe is a sum of e^-(Eo/kT) terms, which is one is dominant becomes
ue (electron potential in solution) = -kT ln [e^-(Eo/kT)] = Eo (dilute solutions)
-- Chemical reaction rates depend on rarely occuring highly energetic states modeled by a Boltzman distribution. The probability of a state above the ground state is
e^-(E/RT)
where
E is the energy of vibrational & rotational levels of
ion-solvent bonds (following a boltzmann distribution)
----------------------------------------------------------------------
Salts
The liquid
metal battery uses molten salts as the electroyte, but what chemically
is a salt? It shows my ignorance of basic chemistry that I don't know.
Of course, the most famous salt is NaCl (sodium cloride) table salt. Sodium
is under hydrogen with one electron in its outer shell, an alkali metal,
and chlorine is a halide with one electron missing in its outer shell,
so are all salts like this?
The molten salt battery article lists several salt electrolytes (lithium fluoride, lithium chloride, lithium bromide, and potassium bromide) and they are in fact all like sodium chloride. Lithium, sodium, and potassium are all alkali metals (col 1) and florine, chlorine, and bromine are all halides (col 7). These salts are all of the form AX (equal numbers of metal and non-metal) and in solid form a rectangular grid of alternating atoms.
Magnesium chloride (MgCl2) and Calcium chloride (CaCl2)I believe I read that when sodium chloride forms it releases a huge amount of energy. The difference in their electronegativity is huge: metal sodium = 0.93V and non-metal chlorine = 3.16V. The resultant product should be very stable (difficult to pull apart). So the first mystery is why is it so conductive? Why does it disassociate into ions when molten? Sadoway mentions lot of salts as possible electrolytes, so it seems that high conductivity is common.
There are also ionic salts of the form AX2 like magnesium chloride and calcium chloride. Magnesium and calcium are both col 2 (two valance electrons) and couples with two chlorine donating one electron to each.
-- The high power capability (or a molten salt battrey) is due to the very high ionic conductivity of the molten salt, which is three orders of magnitude or more greater than that of sulfuric acid in a lead-acid car battery. (Wiki -- Molten salt battery)A molten salt used in electrolysis or as an electrolyte in a high current battery probably needs these characteristics:Wow, three orders of magnitude more conductive! Since a lead acid car battery can deliver 100A with a moderate voltage droop that means molten salt batteries can (potentially) operate at tens of thousands of amps.
Molten salt battery
This is an interesting battery. It can be stored for 50 years without deteriorating, yet be activated quickly. It uses the trick that its electrolye is a molten salt when at 500C or so, but at room temperature the salt is solid and apparently completely non-conductive. Reactive chemicals are used to quickly heat it up and the battery is ready to go. It was invented by the Germans for use in the V2 rocket where the heat of the engine activated it.
Why high conductivity?
-- Is the reaction
involved in dissolving ammonium chloride in water an endothermic reaction
or an exothermic reaction?
Endothermic. The dissolution of most salts is an endothermic process as
is the case when dissolving ammonium chloride in water.
Maybe one explanation for high conductivity of salts is that a lot of heat is used to disassociate the the compound into ions. Most of the salst melt at 500C or higher.Element charts -- ionization, electronegativity, etc
Sodium chloride
By their very
nature metals and non-metals want to join up (& will release energy).
I wouldn't be surprised if the formation of halide salts releases the most
energy of all with the single electron 'transfer' for each atom pair and
the large electronegativity difference.
Can you figure the energy release from electronegativity or ionization energy? (I'm finding it hard online to find the formation equation with redox potentials.)
Ionization
Ionization
energy is the energy (in eV) to remove the 1st electron (from a neutral
atom making a positive ion). There are also nth ionization energy which
refers to the energy required to remove the nth electron (after n-1 have
been removed). Ionization is listed on my periodic chart (lower rt, 1st
Ion Pot) and in web site below.
ionization energy (eV)
electronegativity electron affinity
----------------------------
--------------------- -------------------
hydrogen (H)
13.6 (well known)
2.2
0.75
oxygen (O)
13.6
3.44
1.46
sodium (Na)
5.14
0.93
0.55
chlorine (Cl)
12.97
3.16
3.61
Is there so of a reverse ionization? This would be the energy released to add one electron (to a neutral atom making a negative ion). Yes, it's called Electron Affinity and Wikipedia has a table of values! It's the ionization energy for a (single charge) negative ion! (Electron affinity is on my periodic chart too, lower left)
http://en.wikipedia.org/wiki/Electronegativity
http://en.wikipedia.org/wiki/Electron_affinity_(data_page)
Pauling's electronegativity is calculated from Bond disassociation
energies
http://en.wikipedia.org/wiki/Bond_dissociation_energy
Absolute Electronegativity
In fact Millikan's
measure of attraction between two atoms is the mean of ionization energy
x electron affinity. It's units are eV. This give an absolute voltage.
Electronegativity is a relative scale with hydrogen arbitrarily set at
2.20, so there is a formla to convert Milligan 'electronegativity' to Pauling
electronegativity (usual electronnegativity).
For sodium
chloride
(Na ionization + Cl electron affinity)/2
(5.14 + 3.61)/2 = 4.375 eV
Conversion to Pauling
electronegativity is
pauling difference = 0.187 x (Ei + Eea) + 0.17
= 0.187 (5.14 + 3.61) + 0.17
= 1.64 + 0.17
= 1.81 <=> 2.23 =(3.16 - 0.93)
whoops!
-- The process works slightly differently depending on whether an ion with a positive or a negative electric charge is being produced. A positively-charged ion is produced when an electron bonded to an atom (or molecule) absorbs enough energy to escape from the electric potential barrier that originally confined it, thus breaking the bond and freeing it to move. The amount of energy required is called the ionization potential. A negatively-charged ion is produced when a free electron collides with an atom and is subsequently caught inside the electric potential barrier, releasing any excess energy. (Wiki Ionization)---------------------------------------------------------------------------------------------
Aside on negative heat capacity and negative gravitational energy
Today I was reading Freeman Dyson book where he was discussing how gravitational bound systems, like the sun, are peculiar thermodynamically in that they have negative heat capacity. Heat capacity is the amount of heat required to change the temperature 1C. All objects we are familiar with on earth have positive heat capacity, i.e, they get hotter when energy comes in and cooler as energy goes out. But the gravitational bound sun with its balance of outward thermal pressure and gravitation inward pressure has a negative heat capacity. The sun gets hotter as it radiates away its energy, and the reason is that the energy of gravity is negative.
He makes a telling comment in general the gravitational potential energy will be -2E where the kinetic energy is E, making the total energy of the system [potential energy (-2E) + kinetic energy (+ E) = -E]. In other words in gravitation systems (or atomic orbitals) the total energy (magnitude) is equal to the kinetic energy, but only because in general the potential energy is negative and twice the positive kinetic energy.Hydrogen eV numbersI found this same relationship exploring the energy of electron orbits.
Atomic orbital kinetic/potential relationship is the same as Freeman Dyson talked about for gravitational objects [Potential energy = - 2x kinetic energy] or |potential energy| = |kinetic energy|E potential lost = integral { F dr} (integrate from infinity to r)
School water electrolysis experiment from nasa! that says
the positive electrode is the anode (but this is a power in case!)
http://aquarius.nasa.gov/pdfs/electrolysis.pdf
Internal ionic current flow
-- in a galvanic
cell, contrary to what occurs in an electrolytic cell, no anions flow to
the anode, the internal current being entirely accounted for by the cations
flowing away from it
What's the difference between a galvanic cell and electrolytic cell? Pretty sure an electrolytic cell is used for separating out metal. A discharging battery is referred to as a galvanic cell.
Motion of ions --- diffusion or E field
(anode?) comment
that pargues positive ions move away from negative terminal (metal) electrode
because electrolye looks more negative. really? would imply local
batteries at the electrode interfaces.
-------------------------------------
Calculating voltage
-- The common
non-rechargeable (throwaway) alkaline battery uses a zinc negative electrode,
a manganese dioxide positive electrode, and an aqueous alkaline electrolyte.
The overall battery cell discharge reaction is Zn + MnO2 + H2O = ZnO +
Mn(OH)2. For this chemical reaction the free energy of the reaction products
is lower than the free energy of the reactants, and therefore the reaction
proceeds spontaneously. During battery cell discharge the electrochemical
oxidation reaction at the negative electrode is Zn + 2OH- = ZnO + H2O +
2e-, and the electrochemical reduction reaction at the positive electrode
is MnO2 + 2e- + 2H2O = Mn(OH)2 + 2OH-. One can use the well-known thermodynamic
free energy change of the chemical reaction Zn + MnO2 + H2O = ZnO + Mn(OH)2
along with the Faraday Law to calculate the observed voltage of this cell,
which is 1.5 volts.
-------------------------------------------------------------------------------------------------------------------
Electrode/electrolyte interface
I was thinking
about how the electrode/electrolyte interface must work at the molecular
level on a drive and in short order a picture fell into place which I sketched
up when I got home. The key is to assume that some atoms at the interface
are continually popping off into solution and then look at what forces
are on them as a function of the charge on the electrode. If the ion is
pulled back each time, then there is no net current. An ionic current does
exist if ions at the surface are pushed away from the electrode. Another
possibliity is that popped off ions experience no net force, but ions keeps
coming into solutions, so a flux of them builds up near the electrode,
which causes them to move away by diffusion.
Later I found this college discussion of the interface
(Univ of Sydney, Autstraliz, Physics dept). It looks like it might have
some models.
http://www.physics.usyd.edu.au/super/life_sciences/E/E5.pdf
-- boundary
between dissimilar materials is like having a semiperiable membrane
(semiperiable membrane with ion flow supports a voltage across it)
-- someof
the ion species present can diffuse from one material to the other
metal electrode placed in an ionic solution. interface
with an ionic solution
(concepts are similar to a pn semiconductor interface in a diode)
-- (possible)
chemical reactions at an interface play a role analagous to permiability
in the transfer of ions through a semipermiable membrane
-- copper
electrode in copper sulface electrolyte
Cu <=> Cu2+ + 2e- (<=> means reaction can go either way)
cu2+ ions are the only ion (in this case) that can cross the interface,
so it's like
having a semipermiable membrane that can only pass cu2+
-- if no current
(ion flow) then a double charge layer builds up to give a nernst (equilibrium)
potential across the interface
-- in contrast
consider magnesium in acid
Mg + 2H+ <=> Mg2+ + H2
magnesium atoms combines with two hydrogen ions in solution, release molecule
(H2) of hydrogen gas leaving a Mg2+ ion in solution
in this case two species of ions can cross the interface
interface potential mediates the reactions such that the current flows
carried by H+ and Mg2+ cancel out. The interface acts like a membrane with
different permiabilities for H+ and Mg2+ ions
Cu2+ + 2e- => Cu
+0.34V - Velectrolyte
Zn => Zn2+ + 2e- -0.76V
- Velectrolyte
----------------------------------------------------------------------------------------------------
Battery families
Batteries
roughly fall into three groups: zinc family, lead-acid, and lithium based.
Zinc was used in the first battery in 1800 and has been the foundation
of batteries ever since.
Most small batteries today (button cells, AAA to D cells), while they might have names like Alkaline or silver oxide, are part of the zinc family, and a dissolving zinc anode provides about half of the battery's voltage and power. They typically have a cell voltage of 1.5V and are double electrolyte batteries with a separator. The cathode materials in this family include copper, silver, oxygen, and electrolytes can be acid, basic, or salt.
The lead-acid battery is so common as the car battery that it's not widely recognized (certainly I didn't recognize it) just how extraordinary and unusual is its battery chemistry. It was invented in France 150 years ago (by Gaston Planté), and was the first rechargeable battery, and with over a century of engineering improvements this battery chemistry lives on. The lead-acid battery chemistry is one of a kind, not part of a family, and has several attractive and unusual features.
First, it is rechargeable (technically called a secondary battery).
Second, it has only one electrolyte, which is very uncommon and makes the
battery simple.
Third, it has a high cell voltage (2V) which makes a practical, moderate
voltage battery simpler.
Fourth, as built (before it is charged) the same material (lead sulfate)
is used for both anode
and cathode
This is quit an extraordinary combination of features. Add in that its chemistry is built around a common and inexpensive material (lead) and its electrolyte is also inexpensive (sulfuric acid), and no wonder there are millions built every year.
I haven't really look into lithium batteries. I expect it would be difficult to research lithium battery chemistry at this time, because are new, varied, still under intense development, are more importantly I expect much of the basic lithium chemistry information is just not available because it is proprietary. As an example, I found while writing an essay on hybrid and electric cars that when GM picked a Korean company to provide the lithium ion battery that the Korean battery company instantly took off line the specifications of their higher power lithium cells.
Zinc family overview
Zinc is kind
of an unusual metal. For a metal it comes apart easily. It has a high vapor
pressure, so it can't be used in ultra high vacuum applications. It melts
at a much lower temperature than most metals, and it has a low heat of
fusion (energy of formation). It's in the same periodic group as cadmium
and mercury, which have even lower heats of fusion and melt temperature,
mercury of course, being the only metal whose melt temperature is below
room temperature.
Of course what's relevant for batteries about zinc is that makes a good anode material because it spontaneously comes apart (dissolves) when tugged at by the polar molecules in acids, bases and salts producing a 0.76V potential. (Why it does not dissolve in (polar) water I do not know. Is it as simple as that water is not a conductor and can't supply the neutralizing charge?)
I read the reason why the atomic bonds in these metals are somewhat weak is their electron orbitals have somewhat of a complete flavor because the outer shell is an 's2' pair over a filled 'd10' shell. (This explanation is probably hand waving for a quantum analysis, and many of the metals in the center of the periodic chart have an 's2' outer shell, though not over a filled 'd' shell, because the 'd' shell fills after outer s1 and s2 orbitals.)
1) Volta cell (0.76V) the first battery (invented 1800) and has only one power reaction, zinc dissolving in acid (sulfuric acid).
anode zn => zn2+ + 2e- Eo = 0.76V
2) Daniell battery (1.1V) is zinc and copper in salt electrolytes. Zinc dissolves in (zinc sulfate) salt and copper ions in (copper sulfate) electrolyte plate out (0.35V). First battery with a separator.
anode
zn(s) => zn2+ + 2e-
Eo = 0.76V
cathode
cu2+ + 2e- => cu(s)
Eo = 0.35V
overall
cu2+ + zn(s) => cu(s) + zn2+
Eo = 1.10V
3) Silver oxide battery (1.55V) is zinc with silver oxide in a basic electrolyte. Zinc reacts with the hydroxide ion of a base forming zinc oxide (zinc hydroxide). Silver oxide reacts with water to form pure silver and hydroxide ions. (Zinc with mercuric oxide battery has similar chemistry.)
Zinc is oxidized to form zinc hydroxide in the form of the soluble zincate [Zn(OH)4 2-] ion, which eventually precipitate to form zinc oxide (ZnO).
anode
Zn + 4OH- => Zn(OH)4 2- + 2e-
Eo=1.20V
Zn(OH)4 2- => ZnO + H2O + 2OH-
----------------------------------------------
anode
Zn + 2 OH- => ZnO + H2O + 2 e-
Eo=1.26V
cathode
Ag2O + H2O + 2 e- => 2Ag + 2 OH-
Eo=0.34V
overall
Ag2O + Zn => 2Ag + ZnO
Eo=1.60V
4) Zinc air battery (1.3V) is zinc with oxygen (from the atmosphere) in a basic electrolyte, potassium hydroxide (KOH). Zinc reacts with the hydroxide ion of a base forming zinc oxide (zinc hydroxide). Oxygen (from atmosphere) is reduced (with water) to hydroxide ions.
anode
2Zn + 4 OH- => 2ZnO + 2H2O + 4e- Eo=1.26V
cathode
O2 + 2H2O + 4e- => 4 OH-
Eo=0.40V
overall
O2 + 2Zn => 2ZnO
Ep=1.66V
Competing
anode
Zn + 4OH- => Zn(OH)4 2- + 2e-
Eo=1.20V
cathode
O2 + H2O + 2e- => H2O- + OH-
Eo=-0.076V
2 H2O + 2e- => H2(g) + 2OH-
Eo=-0.83V reduction of water
--------------------------
Wikipedia
anode:
Zn + 4OH => Zn(OH)4 2 + 2e
E0 = 1.25 V
fluid:
Zn(OH)4 2 => ZnO + H2O + 2OH
--------------------------------------------------
Zn + 2OH => ZnO + H2O + 2e-
anode
2Zn + 4OH => 2ZnO +2H2O + 4e-
cathode
O2 + 2H2O + 4e => 4OH
E0 = 0.4 V
overall:
O2 + 2Zn => 2ZnO
E0 = 1.65 V
-----------------------
Clean chemistry
Silver oxide and
zinc air look like they have clean chemistry. OH- is the current carrying
ion through the whole cell, being generated at the cathode and consumed
at the anode. Same with water. Water appears in the equations, but as much
water is generated at the anode as is consumed at the cathode, so the cell
does not consume or export water.
Silver oxide generates (solid) pure silver. Zinc-air (main) product is also a solid, zinc oxide, though there are competing reactions that can generate hydrogen (H2) and zinc hydroxide. I don't know how these products are handled, but the discharge curves (below) for silver oxide and zinc-air are nearly ideal (virtually flat), so clearly the accumulating products in the cells don't interfere with the reactions.
Zinc basic electrolyte cell v-i comparisons
silver, oxygen, manganese
(Duracell makes all three types)
source: http://www1.duracell.com/oem/primary/Zinc/zinc_air_tech.asp
5) Alkaline battery (1.5V nom) is zinc with manganese dioxide in a basic electrolyte (concentrated potassium hydroxide (KOH)). The alkaline manganese dioxide battery is essentially a zinc carbon battery improved by a change of the electrolyte from acidic to basic (adds 0.5V to the zinc reaction), and both zinc and manganese dioxide are powered for low ESR. The alkaline battery physically is like an 'inside out' zinc carbon battery with the zinc in the center surrounded by a ring of MnO (plus graphite) packed in a steel can, which is the cathode electron distributor. No gases are formed in the alkaline battery, unlike the dry cell.
Zinc (powder) reacts with the hydroxide ion of a base forming zinc oxide (and/or zinc hydroxide). Manganese dioxide (MnO2) powder gets slightly reduced with two manganese atoms sharing three oxygen (Mn2O3), thus freeing up one oxygen for OH-.
Note water is consumed at the cathode (making OH-) and regenerated at the anode (where an oxygen leaves OH- to oxidize zinc), so it looks like there is a (slow) flow of water in the cell from inside to outside across the separator! The products ZnO and Mn2O3 slowly build up at the anode and cathode respectively. To keep them from gumming up the works the battery reactants zinc (anode) MnO2 (cathode) are both powered (mixed with their gelled electrolytes).
anode
Zn + 2 OH- => ZnO + H2O + 2 e-
Eo=1.26V
cathode
2 MnO2 + H2O + 2 e- => Mn2O3 + 2 OH-
Eo=0.15V
overall
2 MnO2 + Zn => Mn2O3 + ZnO
Eo=1.41V
competing
cathode
MnO2 + 2H2O + 2e => Mn(OH)2 + 2OH-
Eo= -0.05
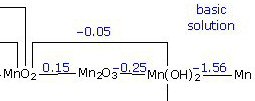
manganese reduction potentials (cropped) from WebElements
Duracell alkaline battery
zinc powder (center) and manganese dioxide powder
(outside)
in basic (KOH) electrolyte
source: http://www1.duracell.com/oem/primary/Zinc/zinc_air_tech.asp
Eveready alkaline app note says, "In general internal resistance will rise during discharge due to the byproducts of active materials." Eveready advertises the alkaline battery as a 1.5V battery, but clearly the battery voltage roll off (at moderate current, see curve below) is substantial, dropping quickly from an initial 1.6V to (1.4V to 1.2V) for about 2/3rd of 'life' and if pushed will give an extra 50% with still lower voltage (1.2V to 0.9V). The discharge curve shows that while ESR goes up with time, it looks like the ESR increase only partially explains the considerable decrease in cell voltage. So if not due to ESR, why does the cell voltage decrease so much?
Looking at the voltage roll off (with time) and the structure of the battery my guess is that the active part of the battery initially is the materials near the separator and that as ZnO and Mn2O3 build up the active region slowly moves backwards. This would lengthen the path of the OH- ions, but shorten the path through the zinc and the MnO2 at the cathode. So is MnO2 a good conductor? This seem unlikely. The answer from the Eveready alkaline battery app note is that cathode material is a "mixture of high purity electrolytic manganese dioxide and a carbon conductor". The same app note describes the anode material as a "gelled mixture of zinc powder and electrolyte". Curiously there is no mention of an electrolyte mixed with the cathode carbon and manganese dioxide.
50 ma discharge with infrequent 500 ma pulses (1 sec
every 12 min)
to measure high current ESR
(source -- http://data.energizer.com/PDFs/alkaline_appman.pdf)
--------------------------------------------------------------------------------------------------------------
Zinc-carbon (flashlight) and LeClanche battery
I found the
Wikipedia and Eveready company descriptions of the chemistry of the standard
zinc carbon (flashlight) battery and its early version ((historical LeClanche
battery) to be incomplete and very confusing. Only when I made the sketch
(below) did things fall into place.
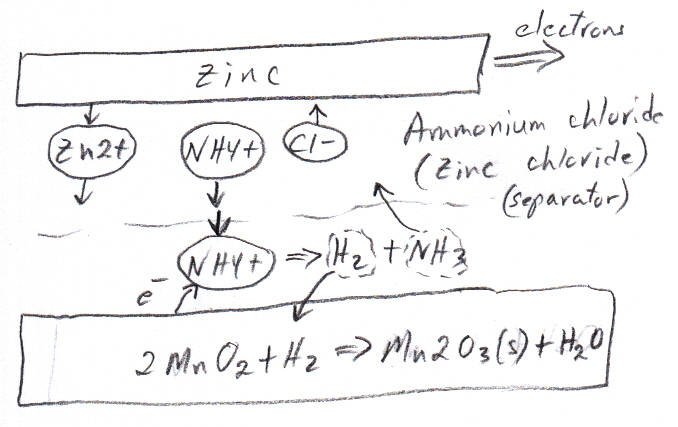
LeClanche and zinc-carbon (flashlight) batteries
Anode electrolyte (LeClanche) -- Ammonium chloride
Anode electrolyte (zinc-carbon) -- Ammonium chloride
+ zinc chloride
The key chemistry can be found in the earlier and simpler LeClanche. The original LeClanche battery consisted of a zinc anode with ammonium chloride (NH4Cl) electrolyte, separator, and manganese oxide (MnO2) cathode. (Eveready calls its standard zinc-carbon (flashlight) battery a LeClanche battery too, which probably indicates it has similar chemistry even though physically it is very different.) The sketch shows the ions on the zinc side of the separator consists of zn2+ (dissolved zinc) and the ammonium chloride salt (NH4Cl), which separates into NH4+ and Cl- ions. The electric field will drive the NH4+ (and maybe the zn2+) across the separator to the cathode, carrying the current across the separator.
At the cathode NH4+ ions are reduced by incoming electrons and separate into two gases hydrogen (H2) and ammonia (NH3). Manganese dioxide (MnO2), which makes up most of the cathode, scoops up the hydrogen gas. I think some of the ammonia is forced either by a pressure build up, or the osmotic effect, through the separator to the anode. There the zn2+ ions (from dissolved zinc metal) and Cl- ions (from the electrolyte) can be considered to be (ionized) zinc chloride, and it scoops up the ammonia that reaches it.
The chemistry
of the (flashlight) zinc carbon is (I think) basically the same as the
LeClanche except that the zinc-carbon has been improved to handle the ammonia
better. It should have more zn2+ and Cl- ions at the anode, since zinc
chloride salt is added to ammonium chloride salt to form the anode electrolyte.
------------------
6a) Original
zinc carbon battery is the LeClanche cell. It's a zinc rod dipped into
ammonium chloride paste outside a linen bag inside of which is a carbon
rod dipped into solid powered manganese dioxide as a depolarizer.
Zn + 2MnO2 + 2NH4Cl => Zn(NH3)2Cl2 + 2MnO2H
EMF about 1.4V
Zn(s) + 2 MnO2(s) + 2 NH4Cl(aq) => ZnCl2 + Mn2O3(s) +
2 NH3(aq) + H2O Wiki
------------------------
6) Zinc carbon
(dry cell battery or standard flashlight) (1.5V) is a zinc can anode lined
with a aqueous paste of ammonium-chloride salt (NH4Cl) and zinc-chloride
salt (ZnCl2 ), and then a separator. Inside and taking up most of the volume
of the cell is a manganese-dioxide ore (MnO2) cathode mixed with powered
carbon (coke, graphite) to increase the electrical conductivity, and a
carbon rod runs up the center to the button +terminal.
The chemistry of this cell is complex and nowhere that I can find completely explained. One reference says it is not fully understood. Here is what I see in the equations: zinc dissolves to zn2+ (typically an acidic reaction, Yes, Eveready says the ammonium chloride and zinc chloride dissolved in water is slightly acidic). Don't know if the zinc ion crosses the separator (probably some does), but it does not show up in the cathode equations. The cathode equations show that NH4+ from the anode must be crossing the separator, so it can carry the current across the separator. The ammonium ion (NH4+) originates from the ammonium chloride salt (NH4Cl) of the anode electrolyte, and after it migrates a(drifts) cross the separator, it is reduced at the cathode to hydrogen gas (H2) and ammonia gas (NH3). The two gases produced must be absorbed within the cell if gas pressure is not to build up.
The manganese dioxide (MnO2) handles the hydrogen and the zinc chloride (ZnCl2) handles the ammonia, both reactions create solids. The manganese dioxide (MnO2) captures the hydrogen and is reduced to dimanganese trioxide (Mn2O3) (a solid plus water). Zinc chloride (ZnCl2) (or the zn2+ ion depending on how you look at it) reacts with ammonia to form solid zinc ammonium chloride. (What is puzzling here is that the zinc chloride is (or seems to be) in the wrong place, on the anode side of the separator.)
Where is the zinc chloride & does it matter?anode Zn(s) => Zn2+(aq) + 2 e- Eo=0.76V
Thinking about it either gas pressure or osmotic pressure is probably going to cause some ammonia to pass from the cathode area through the separator to the anode, and there it will be tied up by the anode side zinc chloride. This will then support a continued flow of ammonia through the separator to the anode.Another possibility is that there may also be zinc chloride in the electrolyte mixed with the MnO2 in the cathode. Many reference are vague on the cathode electrolyte. Some indicate that ZnCl2 is part of the paste that packs the center (cathode), others (like Wikipedia) say ZnLl2 is part of the paste that lines the zinc can. I have never seen an explanation of what the separator is for in this battery. In a lithium ion battery the separator is really just a physical barrier to prevent an anode/cathode short, but in a flashlight battery the zinc (anode) can seems unlikely to short to the center carbon (cathode) rod, so this would indicate that there must be two different electrolytes for the separator to separate. (I suppose it's possible that there is just a concentration difference between the two sides. Controlling where the solid final products are deposited has got to be a key element in the battery design. It may just be that this issue is too deep into the detailed engineering of the cell to be captured in simple battery chemistry descriptions.)
In tables I find
NH3 + H2O => NH3+ + OH-
E0=+ or - 0.28V
NO3- + 10H+ + 8e-=> NH4+ 3 H2O
Eo=0.88V
2H+ + 2e- => H2 (g)
Eo=0V
Variant zinc carbon cell is the 'heavy duty' zinc carbon which has a zinc chloride electrolyte. (Seems like its chemistry must be pretty different because the NH4 ion is gone, but I have not really looked at this cell.)
Zn(s) => Zn2+(aq) + 2 e-
Eo=0.76V
2MnO2(s) + 2H2O + 2e- => 2MnO(OH)(s) + 2OH-
Zn(s) + 2 MnO2(s) + ZnCl2(aq) + 2 H2O => 2 MnO(OH)(s)
+ 2 Zn(OH)Cl(aq)
The zinc (zn2+) chloride (cl-) and hydroxide (OH-) ions join to form Zn(OH)Cl(aq).
Volta
(1800) -- Zinc and copper in sulfuric acid --- single reaction battery
The first
battery 'pile' invented by Alessandro Volta about 1800 had cells of zinc
and copper (he also used zinc and silver) separated by cardboard disks
soaked in sulfuric acid. Wikipedia gives the reactions of Volta's zinc/copper
battery (below), saying the copper does not react. This must mean that
the voltage of each cell is about 0.76V (initially) since this is the potential
of zinc oxidation reaction (zinc dissolving). The hydrogen reaction, hydrogen
ions in the acid being reduced to hydrogen gas at the copper electrode
[2H+ + 2e- => H2 (g)] has a potential of 0V.
zinc
Zn => Zn2+ + 2e-
0.76 V
sulphuric
acid
2H+ + 2e- => H2 (g)
0.0 V
Just two months after Volta invented this battery it was used (by Nicholson and Carlisle) to break apart water (electrolysis) into hydrogen and oxygen, the beginning of electrochemistry.
Volta's paper
I found in an index of classic papers a (reconstructed) version of Volta's 1800 paper and its pile differs somewhat from what is usually put forward as a Volta pile. His preferred way to build a pile is to use disks of zinc and silver separated by pasteboard disks soaked in water (no mention of acid!). His says instead of zinc at the anode (he of course does not use this word) tin also works, but not so well as zinc. Instead of silver for the cathode copper or brass can be used. He does not indicate any difference between them. According to Wikipedia a Volta cell with water as an electrolyte does not undergo a chemical reactions at the cathode, so it probably matters relatively little which of the three cathode metals are used.He describes an experiment putting three 20 cell piles in series, but with the center 20 cell pile flipped in polarity. He then uses his fingers wet with salt water as a voltage detector, running them up and down the pile touching each of the 60 cells and noting how strong the shock is. After a few cells he notes the shock can weakly be felt and strengthens up to the 20th cell, then it gradually decreases again to null at the 40th cell, rising again such that at the 60th cell the shock is the same as at the 20th cell. He understands that current (electric fluid) flows in unbroken loops.
The National
Magnet lab site has a nice Java applet showing ion flows in one of the
simplest possible batteries. Just strips of zinc and copper in a single
electrolyte sulfuric acid are shown powering an LED. (They says this is
basically Volta's 1800 battery.) The zinc dissolves releasing zn2+ into
solution and it stays in solution. H+ from solution is shown migrating
to the copper (forming H2(g) probably at the surface).
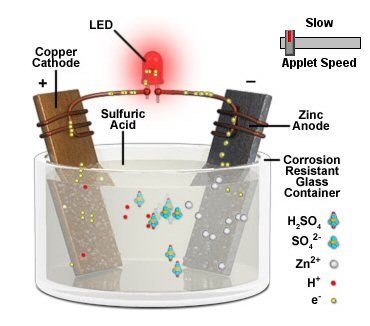
http://www.magnet.fsu.edu/education/tutorials/java/electricalcell/index.html
Thus the current flow near the zinc is all zn2+ ramping down in the electrolyte to all H+ at the copper. In other words the ionic content of the solution changes from 2H+ + SO4- (ionized sulfuric acid) near the copper to zn2+ + SO4- (ionized zinc sulfate) near the zinc. It look like as the current flows the solution changes with (in an ionic sense) zinc replacing the hydrogen in sulfuric acid and forming zinc sulfate.
What about the SO4- ions? The E field will tend to drive them (upward in my sketch) toward the zinc. I put a question mark next to them, because I did not read that they react at the zinc. The applets does show the SO4- ions drifting rightward (toward the zinc electrode), but before they reach it they are look like they are being neutralized by (sort of) joining up zn2+ ions coming off the dissolving zinc.
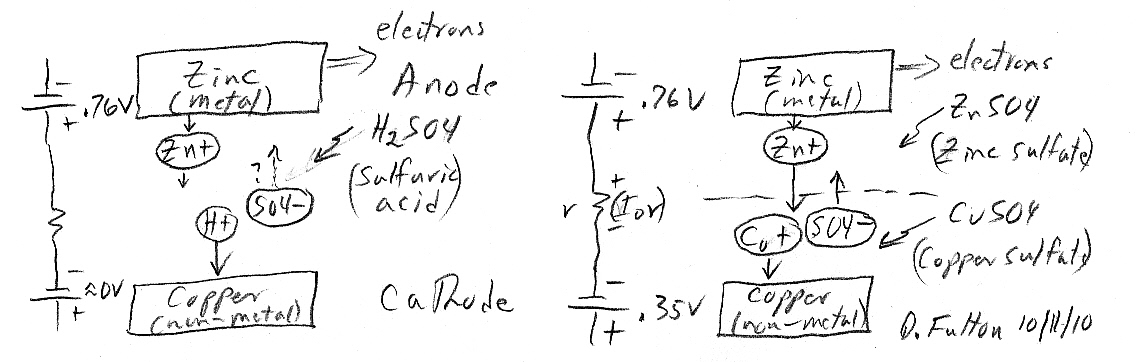
Comparison of Volta cell (left) with Daniell cell
(right)
Daniell double electrolyte chemistry solves Volta's
H2 gas problem and brings copper into play
(zn+ is really zn2+, etc, for simplicity I did not
show valence)
-- any metal you stick in your electrolyte (in this case sulfuric acid) will react by dissolving into positive ions. The difference is in how quickly they dissolve. Zinc dissolves more quickly than copper. So there are more positive ions (depicted as gray particles) in the solution around the zinc than around the copper, and more electrons (depicted here as yellow particles) left behind inside the zinc than in the copper. As a result, there is a potential (voltage) difference between the two.
Good, above is common sense way of explaining why zinc is a stronger metal (oxidizes more easily) than copper. And this seems consistent with the two space large layers in the Sydney lecture (where electrolyte is copper sulfate). But the applet shows no cu+ ions going into solution. Is this right?Volta cell -- stacked zinc, carboard soaked in acid, copper
The crucial element of the invention is that Volta adds a second (copper) electrode to the sulfuric acid + zinc, and because the second electrode is passive (copper doesn't react with the acid), it's at the potential of the acid. The zinc/acid space charge is located between the two electrodes, so the space charge voltage of 0.76V appears on the terminals as an open circuit voltage!
The 'self discharge' spontaneous zinc dissolve reaction into the acid is slowed (I suspect) by using a low concentration of acid and dispersing the acid in the cardboard. When a load is connected across the terminals, it allows the zinc oxidizing (dissolving) reaction to proceed more quickly by bleeding electrons off the zinc anode, which it turn allows more zn2+ ions to go into solution. The beauty part of the 2nd (copper) electrode is that it attracts the H+ acid ions away from the zinc anode to itself, because its voltage is (slightly) negative relative to the acid electrolyte due to (excess) electrons it receives from the external load. At the copper (cathode) electrode the H+ ions are reduced (to hydrogen gas), which because because the H2 bubbles are an insulator and they tend to stick to the copper, gradually reduces the Volta battery voltage and current.
Thus we have at the electrodes an open circuit voltage due to the space charge layer at the zinc/electrolyte interface. With an external load connected we have (positive) ionic flow at both electrodes, so current can flow through the cell with a low impedance and maintain the terminal voltage under load. At the zinc (anode) electrode the more the electrons are bleed off the electrode by the external load the more the zinc can ionize (dissolve) to zn2+ ions. At the copper (cathode) electrode the more the electrons arrive (via the external load) the more the H+ ions can be pulled in from the acid electrolyte and reduced to H2 gas (bubbles). And as Faraday will later formalize there is a one-to-one relationship between electron flow through the load and the oxidiation and reduction reactions at the electrode interfaces.
Volta battery problemsPolarization
It's tough to find info on this, but looking at the electrochemistry it's likely the Volta cell had two major problems. While the battery certainly worked and was useful as a lab tool, it was not a practical battery. One problem was probably self discharge. Zinc is in contact with an acid electrolyte, so open circuit there is going to be some acid H+ (heat generating) current flow back to the zinc electrode allowing the zinc to (slowly) dissolve. The solution to this problem was probably to make a fresh pile or take the battery apart when not in use. My guess is also that battery concentration was kept low (and it was diffused in cardboard) to slow the self discharge reaction.Two the battery slowly dies because non-conducting hydrogen bubbles (from the H+ acid ions) slowly build up and cover the copper cathode. This problem is called polarization. (I am unclear if there is a third problem with zn2+ ions coming to the copper and as they are reduced zinc plating over the copper.) Bottom line is acid, although it allows zinc to dissolve in it and providing ions for conduction, is really a poor choice for an electrolyte because it causes problems at the anode (open circuit) and at the cathode (during discharge).
Volta battery with brine
Again and
again when I think I understand something (in batteries) only to find something
that doesn't fit. After six weeks or so, I think I have a good handle on
the chemisty of Volta's cell. I have been doing all my analysis with sulfuric
acid and copper cathode. Rereading about Volta's cell in Wikipedia and
online I find this Volta also used for his electroyte 'brine' (water saturated
with salt, usually sodium chloride), and although Volta used zinc and copper
in his cell, he found the 'dissimilar metals' that worked best were zinc
and silver.
* Wikipedia
is particularly confusing here. After saying the cardboard was soaked in
either sulfuric acid or brine, they then proceed to analyze the chemistry
only for sulfuric acid. If the postive ion H+ of acid is gone, does the
positive sodium ion replace it? But I find the reduction potential of sodium
is very negative, far more negative than zinc. How can this work? Or is
it the negative clorine ion that handles the current? I am going too have
to get some guidance on salt as an electrolyte. One reference says acid
worked better than brine.
[na+ + e- = na(s) Eo= -2.71V]
* Copper does
appear in the cathode reaction, and copper and silver both do not want
to ionize, so why would it make any difference?
[Ag+ + e- = Ag(s)
Eo= 0.80V]
[cu2+ + 2e- = 2cu(s) Eo= 0.35V]
* Also I focues on a single cell, but Volta stacks alternating disks of zinc and copper. How does this work? I haven't sketched it out, but off the top of my head it seems like the cells potentials alternate and thus will be cancellng.
Solved --- Watching a video of a Voltaic pile being made answered this question. The stack started nickel/cardboard/penny/nickel/cardboard/penny. While nickel and penny are alternated, cardboard goes only betwen nickel and penny. The penny/nickel stack is just the positive and negative terminals of two cells ohmically connected. The voltaic pile is just stack of identical cells in series!
Lemon battery --- demo Volta battery
Stick a copper
coin and a galvanized (zinc coated) nail in a lemon (acidic). Wiki says
voltage is nominally 0.83V @ 150 ua. A YouTube video shows open circuit
reading (multi-meter) of 1.01V for a freshly made lemon battery, but later
tests with multiple lemons show 0.77V to 0.90V/lemon (open circuit). Wiki
give the usual Volta equations for the chemistry. This is higher than 0.76V
zinc oxidize reaction, so how does this work? ( I should try this, buy
a lemon and nail)
Make a Voltaic pile
The anode
of Voltic piles can be zinc, nickel or aluminum. The video below uses both
nickel and aluminum with copper and acid. Video's stack of four nickel/pennies
with acid paper between give 400 mv or 100 mv/cell open circuit. Swapping
aluminum for nickel gives 1.66V or 0.42V/cell. (For some reason these voltages
are far below the reduction potential. In the video it's pretty clear he
is measuring the voltage of multiple (probably four) cells.
ni(s) = ni2+ + 2e-
Eo= 0.26V
zn(s) = zn2+ + 2e-
Eo= 0.76V
al(s) = al3+ + 3e-
Eo= 1.66V
Video: http://www.metacafe.com/watch/3670079/how_to_make_a_battery_from_a_voltaic_pile/
Volta's
flaw and Daniell's fix
From my single zinc
electrode circuit sketch it is immediately obvious there is a big flaw
in Volta's 1800 zinc/copper/acid pile. And it is also clear (though
no one really explains this well), why Daniell in 1836 put zinc in a zinc
solution (zinc sulfate), and copper in a copper solution (copper sulfate).
This fixes the flaw in Volta cell with the result that Daniell cell became
the first practical battery.
-- The Daniell cell consists of a central zinc anode dipped into a porous earthenware pot containing a zinc sulfate solution. The porous pot is, in turn, immersed in a solution of copper sulfate contained in a copper can, which acts as the cell's cathode. The use of a porous barrier allows ions to pass through but keeps the solutions from mixing. Without this barrier, when no current was drawn the copper ions would drift to the zinc anode and undergo reduction without producing a current, which would destroy the battery's life. (Wikipedia -- Daniell cell)Daniell cell --- Much improved Volta battery with two electrolytes
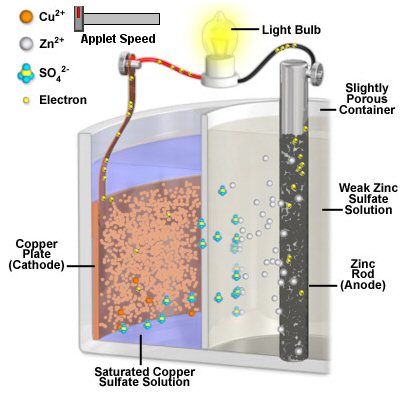
Note zinc sulfate solution starts out weak, because
zn2+ ions build up in solution,
and at the anode zinc dissolving to form zn2+ ions
carries all the cell current.
Copper sulfate solution starts saturated, because cu2+
ions from solution continue to plate out
on the copper cathode, and cu2+ reduction to neutral
copper carries the all cell current to the cathode.
The separator passes only a (reverse flow of) SO4 2-
ions,
and the SO4 2- ions carry all the cell current across
the separator.
(source -- National Magnet lab)
The electrolyte on the copper side is copper sulfate (Cu+ + SO4-), so no H+ of an acid, hence no H2 gas blocking the electrode. The positive ionic current into the copper electrode is now cu+ ions coming out of solution, being reduced to neutral copper (conductor) and plating on the copper electrode. For long battery life the initial copper sulfate solution is 'saturated(and (in another good engineering trick) kept saturated by putting crystals of copper sulfate at the bottom. This provides both a large source of cu+ ions to carry positive current to the copper electrode and a large number of negative SO4- ions to carry (some or most of) the positive current across separator. On the zinc side the zinc electrode is going to dissolve into its electrolyte, so the concentration of its zinc sulfate electrolyte is low ('weak'), so it can absorb lots of zinc.
A little twist is that apparenly the battery is made with weak sulfuric acid (H2SO4) on the zinc side, yet the zinc side electrolyte is usually described (as above) as (weak) zinc sulfate. I think I can make sense of this. Probably what happens is that in the newly made battery the some of the zinc reacts with the H+ of the acid (self discharge), but the reaction stops when the H+ is cleaned out and replaced by zn2+. Thus sulfuric acid turns into zinc sulfate (H2SO4 => ZnSO4). The hydrogen bubbles also have to clear from the zinc.
Self discharge continuesSelf discharge of the cell is prevented by having the only positive ion in the solution touching each electrode be the ion of that electrode. For the zinc electrode with no back flow of positive H+ ions (acids) or cu2+ ions (copper sulfate) possible the zinc is unable to dissolve into the electrolyte, there is no reaction. A circuit model shows no net 'loop' voltage with zinc in zinc sulfate. Each zn2+ reduction absorbs as much energy as a zn2+ oxidation releases. The system comes to equilibrium with (small) amounts of dissolving zinc balanced by (small) amounts of zinc plating out.
Well maybe not all the H+ because on reference says "The contents of the (Daniell) battery had to be dismantled when not used to stop the chemical reactions and conserve the metals." Implying that (self discharge at some level) continued. Or more likely it was cu2+ ions diffusing in from the copper side (through the separator) allowing the zinc to continue to dissolve.Dainiell invented (or worked on) also the gravity cell. The same reference is very specific about the self discharge in the gravity version of the Daniell cell, saying "When the circuit (of the Daniell battery) was opened and left standing, while open the copper ions would diffuse upwards and self discharge onto the zinc anode which resulted in loss of power."
-- Spontaneous (single electrode) reaction occurs when zinc metal is placed in a solution of copper ions.
Zn (s) + Cu+2 (aq) => Zn+2 (aq) + Cu(s)
The zinc oxidizes converting into zinc ions (Zn2+) that dissolve in the copper solution. The negative electrode then attracts (positive) copper ions (Cu2+) that plate out on the zinc (or drop to the bottom of the cell as sediment) as copper atoms. The energy released (in this single electrode reaction) is all dissipated as heat. Note the reaction can continue without a space charge layer forming because downward Zn2+ ionic current is cancelled by upward Cu2+ ionic current.
-- But the energy released can be made to do useful work by a device called, an electrochemical cell. An electrochemical cell is composed to two compartments or half-cells, each composed of an electrode dipped in a solution of electrolyte. These half-cells are designed to contain the oxidation half-reaction and reduction half-reaction separately (as shown). Now there are two electrolytes, zinc is in zinc sulfate and copper is in copper sulfate.
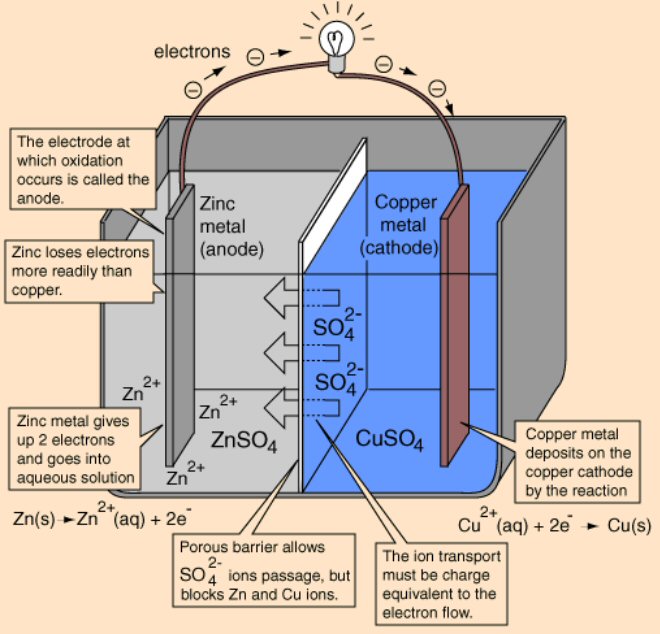
Note Daniell cell separator (ideally) allows only
SO4 2- ions to pass,
so all the cell current at the separator is carried
by SO4 2- ions.
source -- http://hyperphysics.phy-astr.gsu.edu/Hbase/hframe.html
(HyperPhysic site at Georgia State Univ maintained
by Dr. Rod Nave)
Connecting these half cells and letting current flow causes zinc metal to oxidize and go into solution as ions (Zn (s) --> Zn+2 (aq) + 2e-) and copper ions in solution to be reduced and to plate out as copper atoms (Cu+2 (aq) + 2e- --> Cu (s))
Here is the key
The removal
of the Cu2+ ions from the zinc electrolyte eliminates the 'upward' Cu2+
ionic current, so now as zinc dissolves forming a 'downward' Zn2+ current
a space charge layer quickly forms. The space charge forms a nernst E field
that acts to halt the net further dissolution of zinc. (Unstated is why
the electrons released by the zinc (as it ionizes) are unable to go into
solution. Thinking about this H+ can always latch onto H2O in solution
forming they hydronium ion H3O+. Maybe with water there is just no
(equivalent) molecule for the electron to latch onto to form a negative
ion. Maybe this requires chemistry, a solute in the water.)
Does the zinc want to dissolve in the zinc sulfate? If it does, then doesn't copper want to dissolve in copper sulfate? Well then were back to back to back batteries!
Do reduction potential tell the story?-- When metals react with nonmetals, the difference in their electronegativity values is sufficient to justify the generalization that metal atoms will "lose" their valence electrons and that nonmetal atoms will gain these electrons. This generalization is the basis for following guideline. Reactions between metals and nonmetals will usually result in the formation of ionic compounds.
I think (maybe) reduction potential tell the story. Positive redox potential means the reaction is spontaneous.The 'reduction potential' of zinc [Zn2+(aq) + 2 e- => Zn(s)] is negative (-0.76 V), but that means the 'oxidative potential' [Zn(s) => Zn2+(aq) + 2 e-] is positive. Zinc wants to oxidize, to dissolve into zinc ions (in sulfuric acid forming an electrolye with zinc ions in it). The (open circuit) space charge layer that forms has the E field pointing from electrolyte to zinc (pushing the +zinc ions back toward the zinc electrode). The zinc (anode) is thus negative relative to the electrolyte.
But the 'reduction potential' of copper is reversed from zinz [Cu2+(aq) + 2 e- => Cu(s)] is positive (+0.34 V). Unlike zinc the copper electrode does not want to dissolve in a solution with it's own ions. It wants to reduce the +copper ions in solution so they can plate up onto the copper electrode. The (open circuit) space charge that forms with copper (in copper sulfate) points from the copper to the electrolyte (pushing the copper ions in soltion away from the copper electrode. The copper (cathode) electrode is thus positive relative to the electrolyte.
This is consistent with a circuit model being two batteries in series, pointing in the same direction so their potentials add. And it means the voltage of the electrolyte is the node between the batteries, putting the electrolyte potential intermediate between the anode and cathode.
The weakness of this argument is that hydrogen reference is apparently arbitrary, so that makes positive and negative sign of reduction potentials arbitrary. I bet the answer is that with a cell composed of say two metals, both with negative reduction potentials, that the one being forced into being a non-metal must have it's space charge layer flipped. It's cathode is being forced to accept electrons from its more powerful oxidizing companion that will be used to reduce its ions in solution, so open circuit it must form a space charge layer that points into the electrolye, making the non-metal (cathode) electrode positive relative to the electrolyte.
Community college in Hawaii
http://library.kcc.hawaii.edu/external/chemistry/everyday_electro.html
------------------------------------------------------------------------------------------------------
Daniell cell
Zinc (metal)
in zinc sulfate and copper (acting as non-metal) in copper sulfate connected
by salt bridge, which allow zinc+ ions and sulfate- ions to pass.
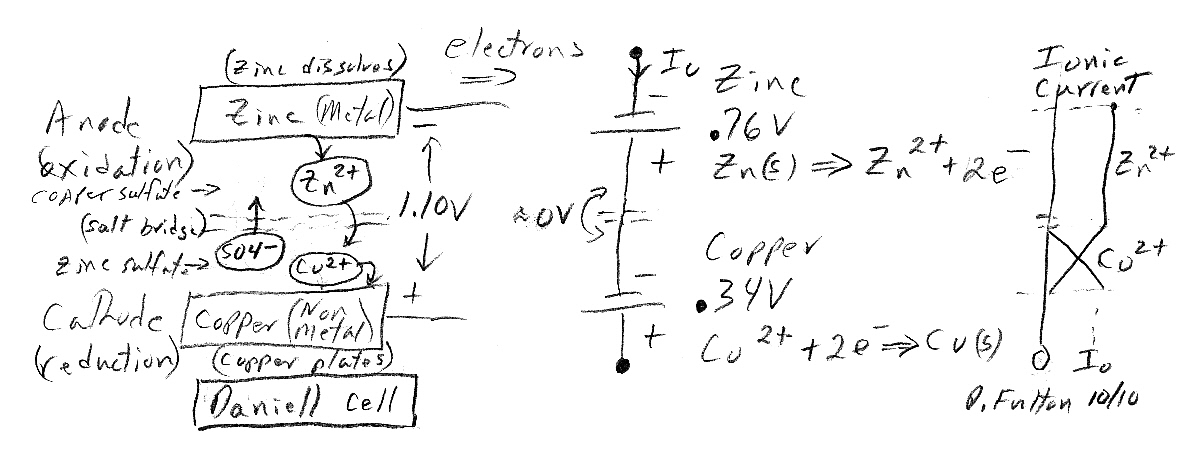
(electrolytes are labelled backwards)
Salt bridge puzzle
All the elementary
texts I read say what a salt bridge does is to conduct ions (between solutions),
but at the same time it keeps two electrolytes from mixing. What the hell
does this mean? On it's face it's a contradiction in terms. The only thing
that defines an electrolyte is its ions, so if they can move through the
salt bridge how are the two electrolytes kept separate.
Salt bridge resistance
Thinking about
this I began to suspect a salt bridge must be a high resistance (or slow
diffusion) type of apparatus. By limiting the ion flow/sec it could be
said it keeps the electrolytes (relatively) separate, the 'relatively'
somehow getting lost in the oversimplified (sloppy) language of elementary
texts. If the purpose of the salt bridge is only to allow a voltage measurement
(where only a very small current need flow), then its resistance can be
quite high. The only thing that matters for an 'open circuit' cell voltage
measurement is that the resistance of the salt bridge by substantially
less than the meter input resistance. But the input resistance of a
standard volt-ohm meter is 10M (and for specialty meters can be much higher).
Hence even a salt bridge with an ohmic resistance of 10k used with a with
a standard 10M input ohm meter allows cell measurements accurate 0.1%.
A google search for 'salt bridge resistance' turns up a 1943 paper (first page). The author is proposing an improved (lower resistance) salt bridge and in his intro he describes the usual 1943 type salt bridge. It's 40 cm long U shaped filled with saturated solution of potassium chloride with two (porous) glass plugs at the ends. Sometimes the solution is jelled to prevent mixing by convection. And reading between the lines it appears that the bridge is just inserted briefly when a measurement is to be made (another point skipped in elementary texts).
The author says the resistance of the standard salt bridge is 7k (6.5k due to the plugs and 500 ohms due to the potassium chloride). His is proposing a 600 ohms version. So here I think I have my answer. A salt bridge is 'high resistance' device suitable only for cell voltage measurements. A check of the electrochemistry chapter in MIT's freshman chemistry book indicates that this is right. Salt bridges are used only to measure open circuit cell voltages current and are not used in 'real' cells.
Zinc
Zinc plays
a central role in elementary electrochemisty and battery lectures.
Zinc in sulfuric acid
Place a zinc
strip in sulfuric acid??, and it dissolves releasing heat. Obviously
this is an exothermic reaction. The ions flows have to balance, so zn+
flows out (dissolving) into solution and leaving behind an e- on
the zinc, and H+ in from the acid electrolyte to the zinc surface
where it picks up the e- left by the dissolved zinc atom and forms H2 gas.
(It may be that exothermic reaction is just zn => zn+ + e- with the inward
H+ flow (to H2) just being driven to maintain charge neutrality.
Gravity (Daniell
type) cell
Thirty years
later (in 1860's) Callaud of France comes up with a variant on the Daniell
cell called the gravity cell. It works better since it has no separator,
and importantly it's easy to built and replenish. It quickly becomes the
staple of the telegraph and stays in service for nearly 100 years. Daniell
initially used an earthenware pot as a separator (later a linen bag), but
here the two electrolytes are kept separate by their different densities,
zinc sulfate floating above the copper sulfate. With the resistance of
the separator gone the battery had a higher usable current.
An interesting twist (according to Wikipedia assembly info) is that zinc sulfate was not put in when the battery was built. Copper sulfate crystal were put on the bottom and then water filled the cell. This probably meant that zinc was bathed initially in dilute copper sulfate. Wikipedia says as current was drawn the zinc sulfate layer would form. But as I see it, with cu2+ ions to provide a back current when the cell was first made, zinc probably spontaneously dissolved (open circuit) until zinc ions swamped the copper ions near the zinc forming a small layer of zinc sulfate.
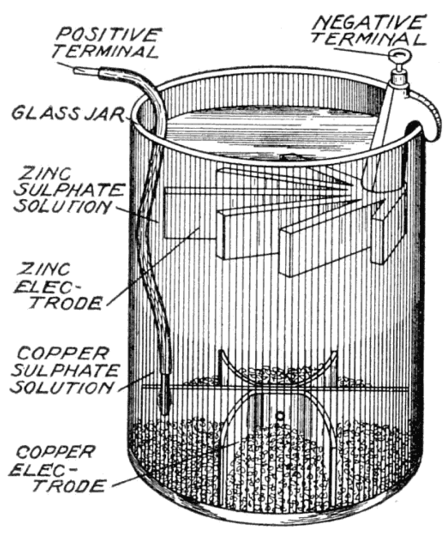
Zinc-copper 'gravity cell' (with Daniell chemistry)
Copper sulfate crystals are at the bottom to keep
the bottom layer cu2+ concentration high (saturated)
Cell is started with water (dilute acid?) and copper
sulfate
Zinc sulfate forms as the cell runs and zinc dissolves
I like this battery. It's physically simple (partially self assembling!) and has simple easy to understand chemistry. It was a staple of the telegraph, going into service in the 1860's and used for almost a century (until the 1950's). It was often built on site, and it's state of charge could be checked visually (probably the fluid separation line moved downward as the zinc anode dissolved and the volume of zinc sulfate grew).
Daniell and gravity cell
Daniell in
a stroke in 1836 fixes both both problems of the volta cell (self discharge
and polarization). He has the brilliant idea to use two electrolytes, each
optimized for its electrode material, which are kept (for the most part)
separate by a barrier that ions can (with some loss of energy) diffuse
through. The result was the first practical battery, and it was used in
the early telegraph.
Daniell's 1836 battery is zinc in zinc sulfate and copper in copper sulfate with a separator between electrolytes. (Later in 1856 the gravity cell version of Daniell's cell eliminates the separator by having the two electrolyses separated due to different densities (& probably surface tension)). There's no significant H+ ions in either electrolyte (zinc sulfate and copper sulfate), so hydrogen gas bubble (polarization) problem is fixed.
A circuit model (see my sketch) shows that the net loop voltage with zinc bathed in a zinc ion electrolyte is zero, so there is no spontaneous dissolve reaction, so the zinc self discharge problem is solved. Yet when oxidized, zinc will still wants to dissolve in this electrode producing a 0.76V space charge. Daniell gets a bonus of a higher cell voltage by bathing the copper (cathode) electrode in a copper rich electrolyte (copper sulfate). Copper ions in solution want to be reduced to plate out [cu2+ + 2e- => cu(s) Eo = +0.35V], so the copper electrode/electrolyte reduction space charge voltage of 0.35V adds to the zinc oxidize (dissolve) voltage of 0.76V making the cell voltage 1.10V.
Space charge open circuit?
Does a space charge
layer develop (as college lecture show). I first wrote 'I doubt it', but
thinking about it a few minutes I am pretty sure the answer is yes. In
fact the zinc electrode develops a 0.76V space charge separation with the
zinc electrode negative to the electrolyte. The E field points from
the electrolyte toward the zinc. This can be viewed as pushing back the
zinc ions that want to diffuse out and causing a cancelling current of
zn2+ ions to be pushed back in to be reduced. This fits nicely with our
circuit model, the space charge is the physical aspect of our circuit model
anode battery.
Note the same thing is true in the case of the copper cathode in a copper solution, but with a critical difference. Cu2+ thermodynamically prefers to plate onto the copper electrode rather than copper electrode to dissolve, so the space charge layer than forms on the copper side is reversed from the zinc side. Here the The space charge E field (0.35V) points from the copper toward the electrolyte. Now the two 'space charge' voltages of the cell add [0.76V + 0.35V = 1.11V] giving us the 1.10V measured externally at the cell terminals.
Zn(s) => Zn2+ + 2e
Eo = +0.76V
Cu2+ + 2e => Cu(s)
Eo = +0.35V
--------------------------
Daniell cell voltage = 1.11 V
What about the arbitrary 0V reference?This applet shows both metal ions are staying on their own side of the separtator and only SO4- passing through the barrier. (Is the barrier SO4- specific? probably to some extent). Is this right? The E field in the electrolyte does act to push the cu+ ions away from the sepatator, but on the zinc side it pushes the zn+ ions toward the separator. Note a daniell cell figure (above) from another source shows exactly this, both zn+ and SO4- passing through the separator (in this case a salt bridge).
The college lecture says in reality the electrolyte is positive relative to both zinc and copper, that both space charges have E fields that point from the electrolyte to the electrodes, that there is added to both circuit model batteries an unknown large voltage that cancels out as measured from the terminals. I still don't know what to make of this argument.On the one hand it seems reasonable physically, but from a circuit model view it leads to an inefficient battery as during discharge large amounts of heat would have to flow between terminals. This just does not happen. So is the space charge layer really there? Wikipedia and other references say the electrolyte voltage is impossible to measure, so who knows if it's there, maybe it's just best to assume it's zero. This is centainly from an engineering viewpoint the best answer and what (in effect) the circuit model does. If it is there, my guess is that it must contain very little energy and is easily dissipated when any small current begins to flow. (If this was true, it would show up as some sort of lowered efficiency for very low currents, but this is something I don't every remember reading about.)
National Magnet lab zinc/copper in sulfuric acid applet:
http://www.magnet.fsu.edu/education/tutorials/java/electricalcell/index.html
-- Zinc is naturally present in water (seawater is 0.6-5 ppb). The World Health Organization stated a legal limit of 5 mg Zn2+/L, which is 5 ppm.
-- Elementary zinc does not react with water molecules. The ion does form a protective, water insoluble zinc hydroxide (Zn(OH)2) layer with dissolved hydroxide ions, according to the following reaction mechanism:
Zn2+ + 2OH- => Zn(OH)2(s)
Zinc reacts with H+ ions, according to the following reaction mechanism:
Zn(s) + 2H+ => Zn2+(aq) + H2(g)
-- Zinc sulfate dissolves in water, forming Zn2+ ions and sulfate ions, SO42-. However, SO42- is a weak base, and so it dissociates water forming HSO4-2 and OH-. Because HSO4- is the conjugate base of a strong acid, it is just a salt and does nothing further.
ZnSO4 + H2O ---> Zn2+ + SO42- + H2O => Zn2+ + HSO4- + OH-
-- The zinc
will react with sulfuric acid forming zinc sulphate dissolved in solution
and releasing hydrogen gas.
Zn(s) + H2SO4(aq) => ZnSO4(aq) + H2(g)
Soluability
-- Most ionic
solids are degraded (dissolved) by polar solvents
-----------------------------------------------------------------------
Silver
oxide battery -- basic electroyte with OH- ionic current
How come button
battery polarities seem to be backwards? What I find is that a silver oxide
cell (like above) is really a zinc/silver oxide cell with a basic electrolyte.
As is usual, zinc is the more active material and is the negative terminal,
but here the zinc is (or is in contact with) the small top electrode. The
silver oxide cathode is in a paste (or liquid) form and fills the lower
half of the metal (nickel plated steel) case.
anode
2Zn + 4 OH- => 2ZnO + 2H2O + 4 e-
cathode
AgO2 + 2H2O + 4 e- => Ag + 4 OH-
combined
2Zn + AgO2 => 2ZnO + Ag
Eo=1.60V
Univ of Cambridge has somewhat different equations for silver oxide battery! (Eo's from WebElement figures below)
anode
Zn + 2 OH- => Zn(OH)2 + 2e-
Eo=1.25V
cathode
Ag2O + H2O + 2e- => 2 Ag + 2 OH-
Eo=0.34V
combined
Zn + Ag2O + H2O => Zn(OH)2 + 2 Ag
Eo=1.60V
Zinc oxidizing in a basic solution, pulling electrons from OH- instead of dissolving as zn+ ions, puts out an extra 1/2 V (1.25V vs 0.76V). If mercury substitutes for silver, the cathode reaction only contributes 0.1V, instead of .34V for silver, so the cell voltage drops 1/4V to 1.35V vs 1.60V for silver oxide.
 .
. 
. 
The zinc metal oxidizes to zinc oxide and silver oxide is reduced to solid silver. Both electrode materials are in gel with their respecitve electrolytes, which are potassium hydroxide or sodium hydroxide (KOH of NaOH). No wonder this is a popular battery. The chemisty is very clever with equal quantities of OH- and water being generated and consumed. OH- can carry the current through the whole cell, because equal quantities are created at the cathode and consumed at the anode.
Metal oxide with zinc family
A number of
battery chemistries involve a metal oxide and zinc in a basic electrolyte.
At the cathode the metal oxide reduces to pure metal, its oxygen combining
with water to form OH-, which carries the current to the anode. At
the anode the zinc oxidizes to zinc hydroxide (Zn(OH)2) consuming the OH-
that were created at the cathode and releasing the electrons to the terminal.
At 1.25V (in a basic solution) the oxidizing zinc provides most of the
cell voltage. Cathode materials can be silver (silver oxide cell, Eo=0.34V),
mercury (mercury oxide cell, Eo=0.1V), or manganese (alkaline, Eo=0.15V).
The basic silver oxide equations apply to a mercury oxide battery [Zn + HgO => ZnO + Hg Eo=1.35V], with a minor twist to (manganese) alkaline cells, and with a somewhat larger twist to zinc-air batteries.
The twist for manganese alkaline batteries is that at the cathode manganese dioxide (MnO2) gets slightly reduced with two manganese atoms sharing three oxygen (instead of four) (Mn2O3), thus freeing up one oxygen for OH-.
cathode 2 MnO2 + H2O + 2 e- => Mn2O3 + 2 OH- Eo=0.15V (WebElements below)
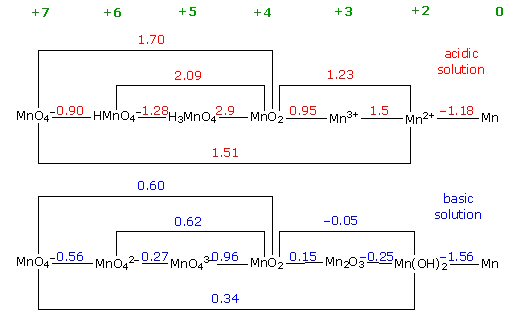
--In order to overcome the chemical reactivity of zinc in alkaline solutions the cells were made with six to eight percent of mercury amalgamated with the zinc to control its corrosion and hydrogen gas formation in the cells.
The zinc air equations are below. The cathode material here is not a metal oxide, but just oxygen that comes in through a hole in battery. In the metal oxides batteries the metal oxide provides the 'extra' oxygen needed to add to water to make OH- ions [O plus H2O plus 2e- makes 2OH-]. The metal oxide at the cathode, having lost its oxygen, becomes a neutral metal. In a zinc air battery cathode there is no metal oxide to reduce. Oxygen just come in from the air and (1/2) O2 combines with the H2O to make 2OH-. Since the cathode material need not be carried around, the energy density per unit of weight of zinc air batteries is high.
anode
Zn + 2OH- => Zn(OH)2 + 2e-
Eo= 1.25V
cathode
1/2 O2 + H2O + 2e - => 2 OH-
Eo= 0.4V
combined
2Zn +O2 +2H2O => 2Zn(OH)2
Eo= 1.65V
-------------------------------------------------------------------------
Zinc - air battery
-- An example
of Metal Air batteries, cells using zinc-air technology are energized only
when atmospheric oxygen is absorbed into the electrolyte through a gas-permeable,
liquid-tight membrane. With the removal of a sealing tab, oxygen from the
air is introduced into the cell. A zinc-air battery usually reaches full
operating voltage within 5 seconds of being unsealed.
The zinc air cell is basically a primary battery however rechargeable designs for high power applications are possible by physically replacing the zinc electrodes.
They use the oxygen content of the air as active mass. The positive electrode (cathode) is a porous body made of carbon with air access. Atmospheric oxygen is reduced at this electrode. The active mass is thus not contained in the electrode but is taken from the surrounding air as it is needed. The initial weight of the battery is reduced accordingly. The negative electrode (anode) consists of zinc. An aqueous solution of potassium hydroxide serves as the electrolyte.
The cell voltage for the chemistry is theoretically capable
1.65 Volts however almost all designs are optimised for less than 1.4 or
1.3 Volts in order to achieve longer lifetimes.
Advantages
High energy density but low power
** In relation to their physical size, Zinc/Air batteries store more energy per unit of weight (in terms of 220 Wh/kg) than almost any other primary type.
High internal resistance which means zinc air batteries must be huge to satisfy high current needs.
The system is well known as a primary battery.
Zinc air button cells are commonly used for watches and
hearing aids.
-------------------------------------------------------------------------------------------------------------------------
Zinc-carbon
(flashlight) battery
The flashlight
battery and classic 6 in high 1.5V dry cell battery is the modern version
(dry version) of the 1866 LeClanche battery. LeClanche's original 1866
battery included all the basic elements (zinc anode, amonium chloride electrolyte,
manganese oxide at the cathode), but his cell was 'wet' (liquid). It has
an acidic amonium chloride electrolyte (NH4Cl) in water and a zinc anode
that dissolves in the electrolyte.
At the cathode the amonium ion (NH4+) is reduced to ammonia gas and hydrogen gas. What is unusual in this battery (and originally confused me) is that the 'thing' being reduced at the cathode is MnO2, which in sketches and descriptions of the battery appears to be the primary cathode material. No, what is reduced, and presumably the reaction that generates about half the cell voltage, is the positive ion (NH4+) of the ammonium chloride electrolyte. The manganese dioxide at the cathode is included to grab the hydrogen, in battery jargon, to depolarize the battery.
anode
Zn(s) => Zn2+ + 2e-
Eo=0.76V
cathode
2 NH4+ + 2e- => 2 NH3(g) + H2(g)
LeClanche and zinc-carbon (flashlight) batteries
Anode electrolyte (LeClanche) -- Ammonium chloride
Anode electrolyte (zinc-carbon) -- Ammonium chloride
+ zinc chloride
Obviously having not one, but two gases, generated inside a battery is bad news, so the zinc-carbon battery has means to degasify (made that up) the cell. Ammonia is tied up by the [zinc + chloride] ions in the electrolyte and a large amount of manganese dioxide is included in the cathode paste to tie up the hydrogen. Chloride ions, from the electrolyte, together with the zinc ions, generated by the anode, (modern zinc-carbon battery also has zinc chloride included in the electrolyte paste) react with amonia gas to make a solid. (one reference says the amonia gas dissolves in the electrolyte).
Zn2+(aq) + 2Cl-(aq) + 2 NH3(g) => Zn(NH3)2Cl2 (s)
The manganese oxide oxidizes the hydrogen gas to water and a more reduced manganese oxide (Mn2O3), a solid. (Manganese oxide functions as a depolarizer.)
2 MnO2(s) + H2(g) => Mn2O3(s) + H2O
Wikipedia gives the combined equation below, which is slightly different from above, for the LeClanche cell. The difference is that instead of Zn(NH3)2Cl2 (s) (whatever this is) the reaction products are shown as zinc chloride plus what looks like dissolved amonia [ZnCl2 + 2 NH3(aq)].
Zn(s) + 2 MnO2(s) + 2 NH4Cl(aq) => Mn2O3(s) + ZnCl2 + 2 NH3(aq) + H2O
and in another article as
Zn(s) + 2MnO2(s) + 2NH4+(aq) => Mn2O3(s) + Zn(NH3)2 2+(aq) + H2O
Univ of Cambridge gives the simplified version (omitting the side reactions), saying "The exact mechanism for this is complicated, and there is still controversy over the exact mechanism." Something curious is that ZnO does not appear in the above equations. (The above equations from Wikipedia have the 'spare' oxygen going to water!)
Zn + 2MnO2 => ZnO + Mn2O3
The battery has a separator, but interestingly the basic electrolyte (amonium chloride) is the same on both sides. The reason for the separator is that on the anode side it's just amonium chloride against the zinc. On the cathode side the amonium chloride electrolyte has mixed in with it manganese dioxide and this combo is (chemically) the cathode. A carbon rod down the center of the cathode paste picks up the electrons (aided by some carbon power mixed into the electrolyte too). The trick of combining an electrolyte and electrode material is used a lot in batteries.
Carbon rod (positive terminal) (surrounded by manganese dioxide (MnO2) plus carbon powder) and zinc can (negative terminal) electrically connected by an electrolyte of water with dissolved zinc-chloride (ZnCl2) and ammonium-chloride (NH4Cl). However, it looks from the figure below that from an electrochemical viewpoint the anode is probably 'manganese oxide' with carbon just a conductor, and later Wikipedia does say the carbon rod is not involved in the electrochemistry. The open circuit voltage is 1.5 V.

There is apparently a paper separator somewhere in
here
(source Wiki Zinc-Carbon battery)
OK the strong oxidizers like oxygen, fluorine and chlorine (all non-metals) in chemical reactions pull in electrons. They have high electronegativity. The non-metal anode pulls in electrons that have flowed from the metal and through the load. That makes the non-metal always the positive terminal and the metal the negative terminal of the battery.
What baffles meI am reading about the zinc-carbon cell in Wikipedia and already I am already confused. The article title is zinc-carbon cell, but Wikipedia says the carbon is not involved in the electrochemistry! The carbon rod is surrounded by manganese dioxide (MnO2). Is this the active anode?
But .... even in this very simple cell the simple metal non-metal thing doesn't seem to work. The anode is (apparently) manganese and the cathode zinc!!When I hear simple battery descriptions what baffles me is why the electrons flow through the outside load anyway. If the electrolyte is just a simple conductor (or is it a diode), why isn't battery short circuited?
Electronegativity
The (open circuit) voltage of a simple two element cell isn't just the difference in electronegativity is it? It can't really be this simple, can it?
Notation: (s) = solid, (ag) = acquous (liquid), (g) = gas
-- The key
reactions of MnO2 in batteries is the one-electron reduction:
MnO2 + e- + H+ => MnO(OH)
This equation is from the Wiki article on manganese dioxide,
but this equation does not appear in the Wiki zinc-carbon cell article!
-- In a zinc-carbon
dry cell, the outer zinc container is the negative terminal. The zinc is
oxidized
(zinc loses an electron) according to the following half-equation.
Zn(s) => Zn2+(aq) + 2 e-
--A graphite
rod surrounded by a powder containing manganese(IV) oxide is the positive
terminal. The manganese dioxide is mixed with carbon powder to increase
the electrical conductivity. The reaction is as follows:
2MnO2(s) + H2(g) => Mn2O3(s) + H2O(l)
-- The H2 comes
from the NH4+(aq):
2NH4+(aq) + 2 e- => H2(g) + 2NH3(aq)
-- NH3 combines with the Zn2+. [Zn2+ + 2NH3(aq) = > Zn(NH3)2 2+(aq)] In this half-reaction, the manganese is reduced from an oxidation state of (+4) to (+3). (I still don't understand what 'oxidation states' are)
-- The overall
reaction in a zinc-carbon cell can be represented as:
Zn(s) + 2MnO2(s) + 2NH4+(aq) => Mn2O3(s) + Zn(NH3)2 2+(aq) + H2O(l)
Getting the reduced form:
Zn(s)
=> Zn2+(aq) + 2 e-
2NH4+(aq) + 2 e- => H2(g) + 2NH3(aq)
2MnO2(s) + H2(g) => Mn2O3(s) + H2O(l)
[Zn2+(aq) + 2NH3(aq) = > Zn(NH3)2 2+(aq)]
----------------------------------------------------------
Zn(s) + 2MnO2(s) + 2NH4+(aq) => Mn2O3(s) + Zn(NH3)2 2+(aq) + H2O(l)
Eveready app note
Zn + 2MnO2 + 2NH4Cl => Mn2O3 + ZnCl2 + 2NH3 + H2O
-- In a "Heavy
Duty" cell. Instead of an electrolyte mixture containing much NH4Cl, it
is largely only ZnCl2 paste. The cathode reaction is thus a little different:
[MnO2 + e- + H+ => MnO(OH)]
MnO2(s) + H2O(l) + e- => MnO(OH)(s) + OH-(aq)
-- as is the overall reaction:
Zn(s) + 2MnO2(s) + ZnCl2(aq)
+ 2 H2O(l) => 2MnO(OH)(s) + 2Zn(OH)Cl(aq)
Eveready app note
4Zn + 8MnO2 + ZnCl2 + 9H2O => 8MnO(OH) + ZnCl2 + 4ZnO + 5H2O
(reduced)
4Zn + 8MnO2 + 4H2O => 8MnO(OH) + 4ZnO
Eveready describes their two zinc-carbon batteries this way:
-- Title of App note is: Eveready Carbon Zinc (Zn/MnO2).So the carbon-zinc cell is really a MnO2-zinc cell.-- The carbon zinc battery uses a zinc anode (zinc is the anode!) and manganese dioxide cathode, and an electrolyte of ammonium chloride (LeClanche battery) and/or zinc chloride (heavy duty models) dissolved in water. (Powered carbon (carbon black) is used in the cathode mix to improve conductivity.)
Really confused-----------------------------------------------------------
After a couple of hours work reading about carbon-zinc (really zinc-MnO2) cells I am really confused. The two types of zinc-MnO2 seem to have totally different equations, but they both generate 1.5V. (Well, LeClanche is 1.55V vs 1.60V for zinc chloride) Also I first though the energy was generated because I saw water coming out, but in the heavy duty type water is consumed!!
-- To remove the hydrogen, a depolarizer is used and this may be mechanical, chemical or electrochemical. Attempts have been made to make simple cells self-depolarizing by roughening the surface of the copper plate to facilitate the detachment of hydrogen bubbles. These attempts have had little success.
-- Chemical depolarization utilizes an oxidizing agent, such as manganese dioxide (e.g. Leclanché cell and Zinc-carbon cell) or nitric acid (e.g. Bunsen cell and Grove cell), to oxidize the hydrogen to water.
-- Electrochemical depolarization exchanges the hydrogen for a metal, such as copper (e.g. Daniell cell), or silver (e.g. Silver-oxide cell).
Here we have
an explanation for some of the chemistry of common batteries.
----------------------------------------------------
Volta pile
So the obvious
problem is how can the Volta pile work, since the zinc is stacked with
cardboard soaked in acid (sulfuric)? Maybe one answer is that a fresh pile
must be made and used quickly before the zinc can dissolve. When current
is drawn, E field across the electrolyte will drive the H+ ions the other
way (to the cathode) flipping the H+ reduction battery around.
Zinc-air
(zinc-oxygen battery
The cathode
reaction of the zinc air battery is quite amazing: Water is reduced to
OH- ions. Water seems so stable, but if you provide it with electrons and
(dissolved) oxygen, the water exothermally ionizes, i.e. the water
combines with dissolved oxygen and electrons to form OH- ions.
The OH- ions generated at the cathode then pass through a separator into a zinc paste anode where the OH- ions exothermally oxidize the zinc to zinc oxide and regenerate the water, which is free to diffuse back across the separator to the cathode. Clearly the current in this battery is all carried by the drift of OH- ions from the cathode to anode.
cathode
O2 + 2H2O + 4e- => 4OH-
Eo = +0.40V
anode
2Zn + 4OH => 2ZnO + 2H2O + 4e Eo
= +1.25V
In the crosssection of a zinc air battery (below) the separator (red) can be seen to be very close to the bottom of the cell (air holes are in the bottom metal). The fact that nearly all the volume of a zinc air battery is anode goes a long way to explaining why this battery has a very high energy density per weight.
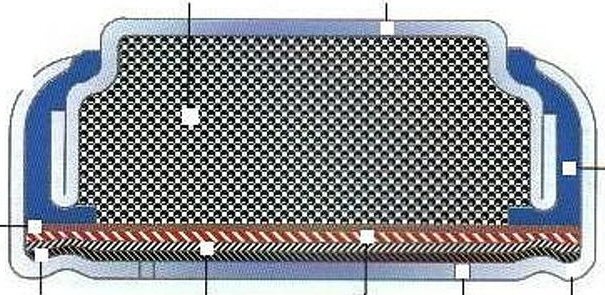
zinc-air battery crosssection (Wikipedia)
above red -- zinc anode (zinc powder in electrolyte)
red -- separator
below red -- cathode (w/air holes for oxygen in metal
bottom)
The electrochemistry of zinc-air batteries (as I guessed) is similar to aluminum air batteries, but with a twist. The zinc hydroxide 'decays' to zinc oxide releasing the water, so the water in the cell is not consumed. Also zinc (I think) does not self oxidize like aluminum, so it's much more practical. As in the aluminum battery, all the current is carried by the flow of OH- ions (generated by oxygen reacting with water and electrons) from cathode to anode. Here's the summary description:
-- In operation, a mass of zinc particles forms a porous anode, which is saturated with an electrolyte. Oxygen from the air reacts at the cathode and form hydroxyl ions which migrate into the zinc paste and form zincate (Zn(OH)4 2-, releasing electrons to travel to the cathode. The zincate decays into zinc oxide and water returns to the electrolyte. The water and hydroxyls from the anode are recycled at the cathode, so the water is not consumed. (Misleadingly worded concerning hydroxyls) The reactions produce a theoretical 1.65 volts, but this is reduced to 1.41.35 V in available cells.(Wikipedia Zinc=air battery)
Anode:
2Zn + 8OH => 2Zn(OH)4 2 + 4e
Eo = +1.25 V
Fluid:
2Zn(OH)4 2 => 2ZnO + 2H2O + 4OH
---------------------------------------------------------
(anode + fluid)
2Zn + 4OH => 2ZnO + 2H2O + 4e
Cathode:
O2 + 2H2O + 4e => 4OH
E0 = +0.4 V
Overall:
2Zn + O2 => 2ZnO
E0 = 1.65 V
Zn from Extended Reduction Tables
This is interesting. Zinc exothermally combines with water to release two
positive ions and two electrons, but potential is less than zn2+ reaction
[zn => zn2+ + 2e- Eo= +0.76 V]. The 3rd reaction listed here
as 1.20V is the anode reaction listed above for the zinc-air battery.
Zn + H2O => ZnOH+ + H+ + 2e-
Eo = +0.50V
Zn + 2OH- => Zn(OH)2 + 2e-
Eo = +1.129
Zn + 4OH- => Zn(OH)4 2- + 2e-
Eo = +1.20V
Zn + 4OH- => ZnO2 2- + 2H2O + 2e-
Eo = +1.22V
-- Metal Air batteries, cells using zinc-air technology are energized only when atmospheric oxygen is absorbed into the electrolyte through a gas-permeable, liquid-tight membrane. With the removal of a sealing tab, oxygen from the air is introduced into the cell. A zinc-air battery usually reaches full operating voltage within 5 seconds of being unsealed.
--An aqueous solution of potassium hydroxide (KOH) serves as the electrolyte. The zinc-air system, when sealed, has excellent shelf life, with a self-discharge rate of only 2 percent per year. In relation to their physical size, Zinc/Air batteries store more energy per unit of weight (in terms of 220 Wh/kg) than almost any other primary type.
Comment -- zinc-copper compared to zinc-air
The zinc-air
is very different from the zinc-copper (Volta/Daniell) cell. In the Volta
cell zinc in acid dissolves releasing positive zinc ions (zn2+) into solution
and
leaving behind its electrons, driven by 0.76V. (In Daniell cell zinc dissolves
in zinc sulfate, also driven by 0.76V.) But in zinc-air the zinc is a base
electrolyte (KOH) and as it goes into solution it reacts with the base
OH- ion yielding (in solution) a negative zinc hydroxide ion (Zn(OH)4 2)
(it later decomposes into zinc oxide, water and hydroxyl ions), driven
by 1.25V. However the zinc hydroxide ion has only half of the charge of
the OH- entering the reaction with the remaining half the electrons
left on the electrode. On the cathode side in the Daniell cell copper ions
in solution want to plate out, driven by +0.35V. In zinc-air oxygen coming
in plus electrons and water react to give (only) hydroxyl ions (OH-), driven
by 0.40V (oxygen gets reduced says one reference).
The zinc air battery has some nice features. The zinc OH- reaction generates almost a half volt more than zinc dissolving does. A continuous flow of OH- ions from cathode to anode (carrying all the current of the cell) can be sustained because equal quantities of OH- are continuously created by the reaction at the cathode and 'consumed' by oxidation at the anode, so OH- concentration in solution remains unchanged. Also conserved is water, the H2O used at the cathode to make OH- ions is all released near the anode (when zinc hydroxyl falls apart).
Aluminum air (oxygen) battery
So if aluminum
wants to dissolve (oxidize) so much more strongly than zinc, how come zinc
is the standard for batteries? Is it economic, or does it have to do with
the flow rates (current)? When I research this, I find aluminum is used
in batteries, and Wikipedia has an article on the 'aluminum battery'. Apparently
current is not the problem, because they could potentially power electric
vehicles. Aluminum anodes have been tested with a several different (non-metal)
cathodes, but apparently there are a lot of practical problems and its
use is restricted to a few military applications.
One basic problem with using aluminum in batteries is that it naturally develops a thin aluminum oxide coating which limits corrosion, but for aluminum to work in a battery the bare aluminum must be exposed, so various tricks and additives are needed to keep the aluminum from oxidizing.
-- Traditional Al-air batteries had a limited shelf life, because the aluminum reacted with the electrolyte and produced hydrogen when the battery was not in use. (One solution was to keep the electrolyte in a tank and assemble the battery when it was ready for use.) Aluminum air (oxygen) battery equation are below (like zinc air this battery has high power per unit weight). (Cell voltage look messed up. Anode oxidation is shown with a negative voltage (-2.71V) which can't be right. It says the total reaction is 2.71V, then says cells generate 1.2V or 0.7V depending on the electrolyte??)
The anode oxidation
half-reaction is
Al + 3OH- => Al(OH)3 + 3e-
Eo = -2.31 V (- ??)
The cathode
reduction half-reaction is
O2 + 2H2O + 4e- => 4OH-
Eo = +0.40 V
At the anode aluminum reacts with hydroxide (base) ion OH- releasing the electron and yielding aluminum hydroxide, which is a major component of aluminum ore. ((Whether it dissolves in the electrolyte or precipitates out as a solid, the article does not say.) At the cathode incoming electrons allow oxygen gas to change water into hydroxide ions. Not only is the aluminum used up (it's the fuel), but the water in the cell is used up too as it (essentially) gets locked up in the aluminum hydroxide It looks like current is carried totally by a flow of OH- ions from cathode to anode. I bet the chemistry of zinc air batteries are similar.
-- Another issue was the formation of aluminum hydroxide as a by-product of the reaction, which would form as a gel-like substance on the anode and reduce the reaction rate. Developments here involve the addition of unique additives to the electrolyte that cause the aluminum hydroxide to form in crystalline powdered form, which fall to the bottom of the cell leaving the reaction surface on the anode clear and active.
-- On the cathode side, delivering the oxygen into the cell has been accomplished through improvements in gas diffusion cathodes, which employ high molecular weight polymers and advanced membrane technology.
Battery amp-hour vs weight
There's a
tight relationship between the total charge a battery can deliver
over its lifetime and the weight of the reactants. Note neither voltage
nor instantaneous current are involved in this relationship, the relationship
is strictly between weight (of consumable reactants) and total delivered
charge (or integrated current).
Faraday figured out these relationships in 1834 by running tests with electrolysis cells. The reason is simple, at each stage inside a battery (anode, cathode and separator too) current can only be carried by ions, charged atoms or molecules. Since battery current flow is DC, each polarity of ion only moves one way. This means that once an ion has delivered its one (or two) electrons to an electrode, it is unable to carry charge again, because it's either it's in the wrong place or has undergone a chemical reaction and is no longer an ion.
Hence to deliver one mole of electrons to the load requires that a new battery start with one mole (or half a mole is the ion is double charged) of (ionizable) reactants at the anode, cathode, and sometime in the electrolyte too for conduction of the current through the separator.
What's a mole of electrons in electrical terms? Ans: It's called a Faraday (F) equal to avogadro's number x charge of an electron.
F = 96.5 x 10^3 coulomb = [6.02 x 10^23 x 1.60 x 10^-19 coulomb]
= 96,500 A for 1 sec
= 96.5 A for thousand sec (17 min) (27 amp-hour)
= 96.5 ma for million sec (11.6 day) (27,000 mah)
= 96.5 ua for billion sec (31.8 year)
Let's work out the weight of reactants needed in a daniell cell to deliver one mole of electrons, which (see above) is roughly 100 ma for 11 days. Current at the anode is carried by zn2+ so we need 1/2 mole of zinc, at the cathode current is carried by cu2+ so we need a 1/2 mole of copper, and current through the separator is (ideally) carried only by the sulfate ion (SO4 2-) so we need a 1/2 mole of sulfate.
anode
1/2 x 65 gram (zinc)
cathode
1/2 x 63 gram (copper)
separator
1/2 x 96 gram (SO4 (32 + 4 x 16))
-----------------------
total 112 gram
For a silver oxide cell the equations are below. For a mole of electrons we need 1/2 mol of zinc, 1/4 mol of silver. OH- and water are not consumed, so from this simple calculation we can't find their weight.
anode
2Zn + 4 OH- => 2ZnO + 2H2O + 4 e- (same in zinc-air)
cathode
AgO2 + 2H2O + 4 e- => Ag + 4 OH-
anode
1/2 x 65 gram (zinc)
cathode
1/4 x 108 gram (silver)
-----------------------
total 60 gram (0.13 lb)
A surprisingly low weight. Think of it this way. A car battery has about 55 Ah rating, so a mole of electrons, which is 27 Ah, from a 1.55V battery, when voltages are accounted for, is roughly 1/16th the power of a car battery. If a car battery is, say 32 lbs, then 1/16th equivalent weight is 2 lbs, so consumables of 0.13 lb (granted different battery type) are only 6% of the equivalent car battery weight.
Let's look up the mah and weight of real silver oxide (button) batteries and scale the 60 grams to the battery's mah rating. Below are two cells about a factor of ten different from the Sony silver oxide family. From the specs it is pretty clear the cell weight and mah rating track closely: a 165 mah cell weights 2.23 gram, while a 16 mah cell weights 0.25 gram.
Silver oxide SR44SW, 11.6 mm dia x 5.40 high, 2.23 gram, 165 mah
(330 ua x 500 hr)
Silver oxide SR521SW, 5.8 mm dia x 2.15 high, 0.25 gram,
16 mah ( 33 ua x 500 hr)
A 165 mah rating in moles is (165 mah/27,000 mah) = 0.0061 mole, so (consumable) reactant weight is (0.0061 mole x 60 gram/mole) = 0.37 gram. This is 16% of the 2.23 gram cell weight. Well it's consistent with the weight, but it does seem kind of low. Some of the difference can be explained by the weight of the electrolyte, case, and maybe other additives. It is also likely that there's a safety factor in the spec, that the consumables are not 100% consumed at the spec mah. So maybe consumables are as low as 16% of the cell weight, but I suspect there is a small error somewhere in this analysis.
The zinc-air battery is very similar to the silver oxide in that it has exactly the same anode equation. There is no stored consumable for the cathode, because it is oxygen from the air. The electrolyte ion OH- separated out from water at the cathode is reconstituted into water at the anode, so no water is consumed. The only consumable for one mole of electrons is 1/2 mole of zinc (1/2 x 65 atomic weight = 32 grams).
Zinc-air PR44, 11.6 mm dia x 5.40 high, 1.77 gram, 344 mah (2.15 ma x 160 hr)
This zinc-air battery is the same size as the large silver oxide above, but it weighs less (1.77 gram vs 2.23 gram). It also has about twice the mah, but not quite twice the power since zinc-air is 1.4V vs 1.55V for silver oxide. Scaling from silver oxide we get the electron mole rating (344 mah/165 mah) x 0.0061 mol = 0.0127 mol, which translates into consumable weight of 0.0127 mol x 32 gram/mol = 0.41 gram. In other words without silver for the cathode this zinc-air button cells does hold about twice the zinc (0.41 gram vs .20 gram) as the same size silver oxide cell. The zinc that will be consumed in a zinc-air battery lifetime is (0.41 gram/1.77 gram) = 23% of the weight of a new cell. (Consumables are in the ballpark of silver oxide, but not quite so low.)
Lead-acid
battery -- Unique single electrolyte battery with high voltage
In
simple terms:
Metal lead (Pb) (partially) oxidizes by pulling a sulfate ion from solution (HSO4-) and joins with the SO4 to form lead sulfate (PbSO4) releasing some energy. The H from the sulfate ion is spit back into solution as H+, The two electrons sent external come from the electron stripped from H (making H+) and from the 'extra' electron carried by the negative ion (HSO4-) pulled from solution.The lead acid (car) battery, the first recharable battery invented (by Plante in 1859), at least conceptually, is amazingly simple. It's a rechargeable battery with a single electrolyte (ideally no separator, but in reality there is one to prevent shorts), and (as a bonus) has a high cell voltage (2V). Idealizing, a fully discharged battery has both plates the same, mostly lead sulfate (PbSO4), and the electrolyte is (mostly) water. During charging lead sulfate (PbSO4) at the negative terminal is reduced to pure lead lead metal (Pb) and at the positive terminal lead sulfate (PbSO4) is oxidized to (non-metal) lead oxide (PbSO2) with both plates throwing HSO4- ions and (net) H+ ions into solution, turning the water into sulfuric acid.The non-metal lead oxide (PbO2) is reduced by pulling a sulfate ion from solution (HSO4-), replacing O2 with SO4 to form lead sulfate (PbSO4) and freeing two oxygen (O). Three H+ are also pulled from solution and combined with H from the sulfate ion, the two freed oxygen, and three electrons form water, which releases a lot of energy. Two of the needed three electrons come in from the outside and the third electron is stripped from HSO4- ion.
When the battery
discharges, both the Pb and PbO2 electrodes pull sulfate ions (HSO4-) from
solution and plate up their electrodes with lead sulfate (PbSO4). At the
negative terminal lead (Pb) oxidizes to lead sulfate (PbSO4) and
at the positive terminal lead oxide (PbO2) is reduced to lead sulfate
(PbSO4) with both plates pulling HSO4- ions from solution and pulling
out (net) H+ ions too. Most of the battery energy comes (I suspect) from
the making of water at the negative terminal by combining the H+ (mostly
from solution) the freed oxygen (from reduced PbO2) and the reducing electrons.
[1/2 O2 + 2H+ + 2e- => H2O
Eo=1.23V]
Discharge (reverse the arrow for charge)
anode
Pb + HSO4- => PbSO4 + H+ +2e-
Eo=0.356V
cathode
PbO2 + HSO4- + 3H+ + 2e- => PbSO4 + 2H2O
Eo=1.685V
-------------------------------------------------------------
overall
PbO2 + Pb + 2HSO4- + 2H+ => 2PbSO4 + 2H2O
Eo= 2.041V
Ed Weisstein's World of Chemistry has these equations
(different sulfuric acid ions)
Hyperphysics site shows sulfuric acid in lead acids batteries
dissociates into [H2SO4 => SO4 2- + 2H+]
anode
Pb + SO4 2- => PbSO4 + +2e-
Eo=0.356V
cathode
PbO2 + SO4 2- + 4H+ + 2e- => PbSO4 + 2H2O
Eo=1.685V
-------------------------------------------------------------
overall
PbO2 + Pb + 2SO4 2- + 4H+ => 2PbSO4 + 2H2O
Eo= 2.041V
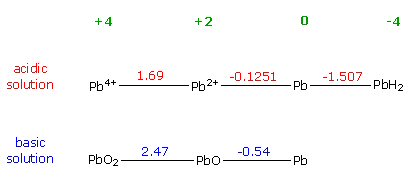
parsing --- Lead (weakly) oxides into a positive ion.
Just as Pb2+ enters solution, it merges with negative SO4 2- ion and plates
out as solid lead sulfate (presumably releasing 0.231V in formation energy).
anode
Pb => Pb2+ + 2e-
Eo=0.125V (WebElements)
Pb2+ + SO4 2- => PbSO4
(Eo=0.231V)
combined anode
Pb + SO4 2- => PbSO4 + +2e-
Eo=0.356V
Sulfuric acid
Seeing different equations given for lead acid batteries I checked Wikipedia to see how sulfuric acid ionizes. Turns out that pure sulfuric acid in equilibrium ionizes into four or five different ions, but more relevant to lead-acid batteries is the ionization of sulfuric acid in water (sulfuric acid concentration in a charged battery is 25-30%). When acid and water are mixed, Wikipedia gives these equations: (hydronium ion, (H3O+) is usually written shorthand as H+):H2SO4 + H2O => H3O+ + HSO4-
HSO4- + H2O => H3O+ + SO4 2-What this says is that sulfuric acid ionizes into [H+ SO4-], but at least some of the HSO4- further ionized to [H+ + SO4 2-]. No equilibrium constants are given, so it's probably reasonable to assume that sulfuric acid contains all three ions: H+, HSO4-, SO4 2-
From the looking at the ions into and out of solution at both electrodes I was able to draw the ionic currents of the battery. Consistent with an idealized battery I sketched the distribution as linear, but who knows what the shape is. The flows are hard constrained at the electrodes and everywhere between the plates the sum of H+ ionic current and HSO4- ionic current (moving opposite) must add to the terminal current (here sketched as 2Io).
How to the ions move?
How to the
ions move? Presumably by a mixture of diffusion and E field, but I suspect
in this battery mostly due to E field. Initially when getting into batteries
I looked at the terminal voltage and drew an interior E field from the
positive terminal to the negative terminal. But this is not (in
any battery) the way to figure the electrolyte E field. The terminal voltage
is nulled by the two interface batteries, so ideally the voltage
across the electrolyte is zero. In fact its resistance is ohmic
with the resistance proportional to the net charge (number of ions), so
the local E is along the direction of (positive) current flow [ delta V
= I x r, where r is the electrolyte ohmic resistance].
I show an 'r' representing the small ohmic resistance of the electrolyte (which actually goes up as the battery discharges and the ions are removed). In discharge (left) the voltage across 'r' is positive on top and negative on the bottom (aligned with the direction of positive current flow), so 3 of the 4 ion flows are push by local E field in the directions they move, as shown by the sketch arrows. But notice the HSO4- ion entering the reactions at the metal (top left). It must move (by diffusion presumably) against the E field in the electrolyte (both in discharge and charge). My guess is battery designers have tricks here to keep this ionic flow adequate. (Watch for this.) This is the kind of subtlety I never see discussed.
A discussion in Wikipedia (lead-acid battery) about 'quick charging' a car battery throws some light on whether ions move due to E field or by diffusion. When a fully discharged battery is recharged quickly ('quick charge'), the battery terminal voltage rises to normal, but the electrolyte ionic charge is 'mostly near the electrodes', so if the battery is left for hours the high conconcentration of ions near the electrodes diffuse away throughout the electrolyte causing the battery voltage to drop. Here's what I think this may mean: Along the pathways between plates the E field drives the charge, but there is substantial additional electrolyte within the battery away from E field, and here a (very) slow diffusion process keeps the ionic concentration in sync with the charge of the battery.
Interestingly 'fast charge' effect works in reverse too. If a car battery is drained by a few minutes of hard cranking, much of the charge lost has come from near the electrodes. If left to rest for a while, the ions in the electrolyte will redistribute (by diffusion) and the battery will partially recover.Lead-acid battery --- early
Charged
one electrode of lead (Pb) and one electrode of lead-oxide (PbO2)
with an electrolyte of sulfuric acid (H2SO4)
Discharged
both electrodes lead sulfate (PbSO4) and electrolyte close to pure water
Below are charging
reaction (discharging are reverse). At anode PbSO4 (driven by 1.685V) picks
up water and the plate converts to PbO2 and the sulfate is released throwing
off one molecule of sulfuric acid (HSO4-). As (positive) current goes into
the anode 2 electrons come out for every molecule of PbSO4 made
and 3 H3O+ ions are released, which charge balances with the one HSO4-
made.
At the cathode
PbSO4 (driven by 0.356V) picks up one of the H3O+ made at the anode and
the plate converts to (pure) lead (Pb) with the thrown off sulfate making
another molecule of sulfuric acid (HSO4 made.
During charging for two positive charges that flow into the anode the anode 'spits out' into the electrolyte four ions (3H3O+ and HSO4-) with a net charge of +2. At the cathode the (external) two charge flow consists of one H3O+ absorbed (from electrolyte) and one HSO4- spit out (into the electrolyte).
I'm not sure how to think about how charge is carried in the electrolyte, but it's probably best to think of the battery as a capacitor with the ions being the charge. But it's weirdly asymmetrical with the anode, which is strongly driven, throwing off four ions (three positive (H3O+) and one negative (HSO4-), while the weakly driven cathode only throws off one ion (one negative ion (HSO4-) while absorbing another (one positive ion (H3O+)).Note the electrolyte in a charged battery ends up neutral. For each pair of PbSO4 molecules created (one at each plate), requiring two charges to flow through the battery, 2HSO4- and 2H3O+ (net) are thrown into the electrolyte. Note water is also consumed as the battery is charged (5H2O in and one H2O out) with the water broken up to make the ions in liquid H3O+ and HSO4-.
In other words when charging the battery each (external) charge flowing through the battery 'makes' a pair of ions (one positive and one negative) in the electrolyte! And of course during discharge the reverse occurs: Two ions (one positive and one negative) disappear from the electrolyte solution (as the plates change from Pb and PbO2 => PbSO4) for each external charge the battery delivers.Anode (oxidation):
Cathode (reduction):
PbSO4(s) + H3O+(aq) +2e- => Pb(s) + HSO4-(aq) + H2O(l)
Eo = -0.356V
Wiki article on Lead Dioxide gives the overal leak-acid
battery equation as:
Pb + PbO2 + 2HSO4- + 2H+ =>2PbSO4 + 2H2O
E = +2.05V
equivalent to above with
2H3O+ => 2H2O + 2H+
Recharable lead-acid battery
When uncharged
it consists of two lead sulfate plates in dilute sulfuric acid. When charging
current flows it one terminal is reduced to lead, the other terminal oxidizes
to lead dioxide and the dilute sulfuric acid becomes concentrated sulfuric
acid as H+ and SO4- ions enter from the reducing and oxidizing plates.
The reactions then run in reverse as the battery discharges, the (negative)
lead terminal (anode) oxidzing to lead sulfate and the (positive) lead
dioxide terminal (cathode) reducing to lead sulfate. As a bonus, the cell
voltage is quite high at 2.0V.
---------------------------------------------------------------------------------------------------
Lithium ion batteries
It is common
in batteries for some element (like zinc) at the anode to oxidize and dissolve
as ions (zn2+) into solution, and at the cathode for a metal oxide (like
MnO2) to be reduced (lower oxidation state). A lithium ion battery works
something like this with an interesting twist.
In a lithium ion battery oxidation at the anode releases lithium ions (Li+) from a carbon matrix into solution where they drift all the way to the cathode (carrying the current). At the cathode reduction of a metal oxide (like CoO2) causes it to take in (and store) the lithium ions from solution. The twist is the lithium ions within both electrodes are not really changed, they remain Li+. They are just reversibly stored (by what Wikipedia calls a phase change), and when current flow reverses (cell is charged) Li+ ions leave the metal oxide and rejoin the carbon matrix.
Ionically lithium ion batteries are very simple. It is a single electrolyte system with one ion (Li+) carrying all the current. At the anode Li+ ions (embedded in graphite a conducting matrix) diffuses out, drift to the cathode, and at the cathode they diffuse into the crystals of a metal oxide (like cobalt oxide or manganese oxide).
A lithium ion battery has a (very high) nominal cell voltage of 3.6V (3.7V for lithium cobalt). I can't find half reaction Eo's, but I think the high oxidation voltage of lithium provides most of the voltage (2.9V) and reduction of the oxidation number (4 to 3) of the metal oxide provides the remaining voltage. Charging just reverses the ion flow, Li+ diffuses out of the metal oxide and back into the graphite. Non-rechargable lithium batteries work pretty much the same way except that the anode is simple metallic lithium which dissolves.
The most unusual aspect of rechargeable lithium ion batteries work is how the lithium 'combines' with the anode and cathode materials. It's not (apparently) a real chemical reaction. The anode and cathode materials are materials with layered (or open) crystal structure that allow the small lithium atom to readily diffuse in and out (solid diffusion) forming lithium (ion) layers a few atoms thick. Diffusing in is called intercalation (diffusing out deintercalation), and this process has been well studied for almost 25 years.
The graphite with lithium atoms inside (anode) is written LiC6 in the equations, but really it's a layered ionic structure. The lithium atoms lose their one 2s valence electron to the carbon atoms (so essentially lithium at the anode remains Li+), and the lost electrons are non-localized, so the graphite becomes a good conductor. I read a paper that showed that lithium dissolving out of the graphite into the electrolyte has an oxidation potential only about a 0.1V less than dissolving from pure metallic lithium.
The metal oxides cathode materials are also layered (or an open 3d matrix) materials that allow Li+ ions to diffuse in and out. Wikipedia says that in a cobalt oxide lithium ion battery the cobalt oxide at the cathode has its oxidation state reduced from Co4+ to Co3+ (missing four electrons to missing three electrons) during discharge. Below is the cobalt reduction potential (from WebElements) showing positive reduction potentials (0.7V to 1.4V) from +4 oxidation state of CoO2 to +3 oxidation state, so maybe the contribution to the cell voltage of 0.7V or so is from the metal oxide reduction, and somehow the lithium is just sucked in. (Manganese oxide shows a similar pattern with a positive reduction voltage for a reduction in oxidation state from MnO4 or MnO2).
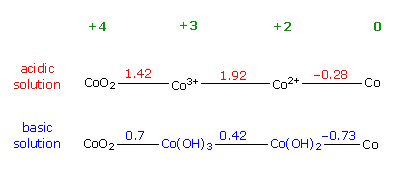
source --- WebElements
The implication is that the metal oxide crystals absorb the incoming electrons, so the lithium remains as Li+ ions between the metal oxide layers, just like at the anode.
Just how does the cathode work?
What I don't
understand (and have yet to see explained) is why lithium ions so want
to diffuse into the metal oxide that it is an exothothermic reaction,
but here is a guess. My guess is that the metal oxide structure somehow
ties up and stabilizes lithium valence electron shifting the dynamics (Fermi
energy bands) such that lithium 'wants' its valence electrons to fit in.
At the same time it can't immobilize the valence electrons, because the
metal oxide with or without a lot of lithium in it needs to be a good conductor.
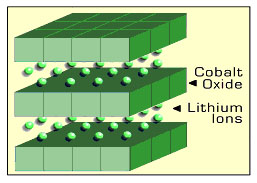 .
. 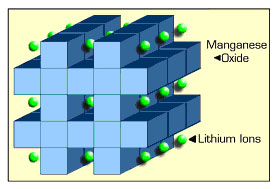
Lithium (Li+ ions) 'intercalates' into cobalt oxide
and manganese oxide crystals,
which are used as cathode materials in lithium ion
batteries.
(source -- http://batteryuniversity.com/learn/article/the_high_power_lithium_ion)
A123 Systems technology, build on work at MIT, is focused on improving the cathode. A123 uses nano size iron phosphate oxide cathode particles in their cathode to greatly expand the surface area, which should made intercalation much more effective by shortening the diffusion path, thus increasing the conductivity of the anode resulting in lower cell ESR and a higher cell current.
anode
(layered) graphite
cathode
(open crystal) metal oxide (manganese oxide in 1st gen electric cars)
electrolyte lithium
salt (typically LiPF6)
Water cannot be part of the electrolyte as lithium (group 1 element)
reacts strongly with water yielding hydrogen gas, so the lithium
salt is dissolved in a non-aqueous organic solvent
At the anode the lithium atoms diffuse out of the graphite into the LiPF6 (lithium hexa-fluoro-phosphate) electrolyte as Li+ ions. The Li+ drift to the cathode and diffuse into the (open, see figure) metal oxide (another intercalation). This is kind of surprising as lithium atoms thermodynamically want to diffuse out of the graphite and into the metal oxide! There's also a separator in the battery, but I don't know what it does as there is only one electrolyte. Perhaps it is to block the PF6- ions from reaching the anode.
Flamability
The battery
electrolyte is an 'organic' solvent with a lithium salt (LiPF6) disolved
in it. 'Organic' means the electrolyte in a hydrocarbon, so it's
flamable, it burns! Worse with all the press about the Boeing battery fires
I read the lithium cobalt oxide coating on the cathode breaks down when
it gets hot releasing oxygen, so the cell can burn internally without the
need for outside oxygen.
Boeing 787 cobalt lithium ion battery (3/8/13)
Investigation into
the fires in the 64lb, 30V, 65Ahr, 8 cell cobalt lithium ion battery (8
x 3.7V = 29.6V) used in the Boeing 787 Dreamliner plane have provided some
details as to how a large cobalt lithium ion battery is made.
anode
copper foil coated in carbon (graphite)
cathode
aluminum foil coated in lithium cobalt compound
The very large (6 lb) cell used in the Boeing battery is model LVP65 made by the Japanese company GS Yuasa. Here's the GS Yuasa spec sheet for the cell. It has a wide temp spec of 0 - 149 F and (for lithium oxide) a high discharge current of 5C (5 x 65A = 325A), however the cell would have 79W of ohmic dissipation at that current. Maximum charge current is 1C (65A). Curves show an ESR of [0.15V/(250A - 50A) = 0.75 mohm], which translates into 30W of loss @ 200A, 7.5W @ 100A and 3.2W @ 65A. (When the Boeing battery problem arose, I made a local copy of the data sheet figuring out that at some point as the Boeing battery problem dragged on, it would disappear from the GS Yuasa web site, which now seems to have happened.)
-- There are four phosphate formulations from which to choose a cathode material: iron, cobalt, nickel and manganese. Manganese is essentially a voltage compromise between the other three. Lithium iron operates the lowest at about 3.2 to 3.3V, whereas lithium manganese operate s little higher (3.5 to 3.6V).
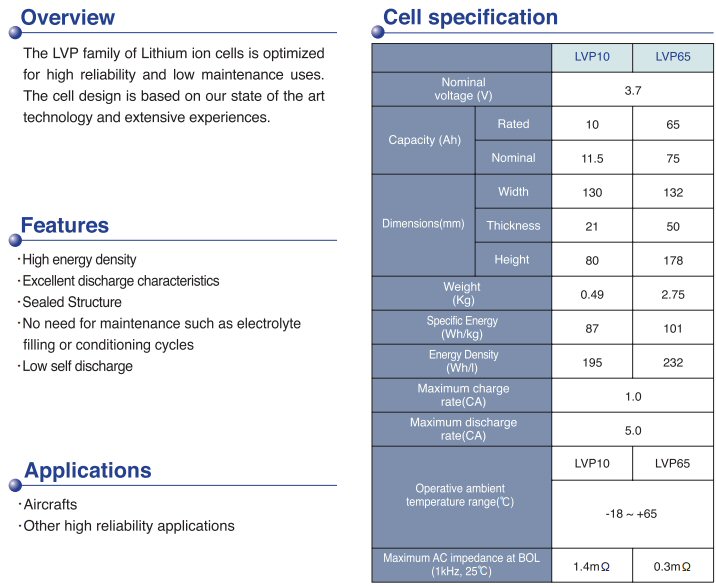
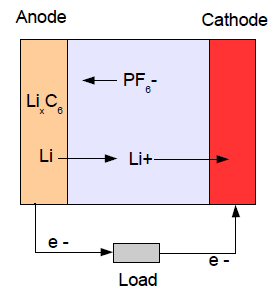
(Note the anode and cathode labels look like they
are reversed in this figure,
lithium metal oxide is normally the cathode)
This figure assumes lithium carbonate (Li2CO3)
PF6- (hexafluorophosphate) is the negative ion of
the electrolyte salt LiPF6 => (Li+ PF6-)
(source --- http://www.meridian-int-res.com/Projects/How_Much_Lithium_Per_Battery.pdf)
It appears that like a lead acid battery a (high power) lithium ion cell is a single electrolyte cell with no separator. (No, there is a separator, but only one electrolyte!) The electrolyte in a lithium ion battery (is usually) is a salt of lithium (LiPF6). LiPF6 is lithium, phosphorus, fluorine, which ionized to Li+ and PF6-. As the figure above shows current at the anode is carried by both Li+ oxidizing and PF6- coming in from solution, but (I am pretty sure) all the anode power (& most of the battery power) is coming from the Li+ oxidizing and going into solution. Battery designers work to minimize the current carried by PF6-, but still (according to the paper I am reading) almost 2/3rd of the current flow at the anode is due to PF6-. (Somehow almost no other reference mentions this, so I doubt it. Is maybe the purpose of the separator to block PF6- from the anode?)
A paper claims that the effect of the PF6- ionic current is that Li+ concentration near the anode is high and is low near the cathode. The paper says this Li gradient across the cell produces a counter voltage across the electrolyte that opposes the cell voltage and dissipates some of the battery energy. In other words the high Li concentration near the anode and low concentration near the cathode produces an E field across the electrolyte with the positive terminal near the anode (negative cell terminal). I think this effect is called the 'concentration polarization'.
Wow, wait a minute these equations in the paper can't be right
anode
Li => Li+ + e-
Eo=+3.0V
cathode
Li + e- => Li
Eo=-3.0V
Wikipedia gives the equations of a lithium ion battery (for a cobalt oxide cathode) as
anode
LiC6 => Li+ + e- + 6C (LiC6 is lithium intercalated into
carbon)
cathode CoO2
+ Li+ + e- => LiCoO2
(lithium cobalt oxide)
For electric vehicles cobalt is too expensive and has a risk of explosion, so the (potential) cathodes are LiFePO4 (A123 Systems) and lithium manganese. Both GM Volt and Nissan Leaf are using lithium manganese, but lithium iron is heavily funded and A123 batteries are going to be used in the Fisher Karma extended range electric. The cathode reaction is basically the same with metal oxides with lithium reversibly squeezing into the metal phosphate 'crystal' as the metal phosphate group is reduced and exiting as it is oxidized. Wiki describes the intertion/extraction of lithium as a 'phase change' of the metal phosphate.'Intercalation' or how lithium combines with a layered material like graphite
Cobalt vs manganese
A lithium
ion battery with a cobalt oxide cathode provides twice the energy density
of a cell using manganese oxide. In practice this means a standard 18650
cell (cylindrical 18 mm dia x 65 mm height often used in laptops) is rated
2.4 Ahr if it uses cobalt, and 1.35 Ahr (out of date?) if it uses manganese.
Laptops, cameras and cell phones use cobalt type cells for long life. Manganese
provides better cathode conductivity so the cell has lower ESR and higher
discharge current and is thermally more stable. Both the GM Volt and Nissan
Leaf electric cars have lithium ion batteries with manganese oxide cathodes.
Lithium battery history
1970's first
primary lithium batteries has metallic lithium anodes. Attempts to make
this type of cell rechargeable failed because dissolved lithium metal did
not grow back cleanly (dendrites form). This problem was solved (and commercialized
in 1981 by Sony) by replacing the lithium metallic anode with a graphite
anode into which lithium ions can easily diffuse. This is the (rechargeable)
lithium ion battery. A phenomena called 'intercalation' allow the small
lithium ions to easily diffuse in and out of the graphite crystal to the
electrolyte. I read a paper showing that this type of anode has an oxidation
potential only 0.1V lower than a metallic lithium anode (about 3V), but
of course, the anode carbon adds volume and weight, so energy density of
the cell is lowered.
The intercalated lithium graphite anode works well, has been in use since 1997 and is considered fairly mature technology. The lithium ions donate electrons to the graphite making it quite conductive. Most of the research and development in lithium ion batteries is focused on improving the intercalated metal oxide cathode, specifically increasing its conductivity to raise the cell current. I see the discharge rates between lithium ion batteries with different cathode metals differs hugely (x35) indicating that the ESR of the cell is to a large degree controlled by the conductivity of the cathode. A chart at the Battery University site (Canadian battery test house) shows Li-cobalt at 1C, Li-manganese at 10C (40C pulse), and Li-phosphate at 35C continuous. Li-phosphate is A123 system's lithium iron phosphate technology. A123 gets the cathode conductivity up by using nano technology to make the metal oxide particles very small (100 nm in dia), hence huge surface area to the electrolyte and short diffusion paths.
Separator (minor) mystery
Most all descriptions
of lithium ion batteries indicate that it has a separator (thin polymer
film), but they never say what it does! One guess I have is it's there
to block PF6- ions of the electrolyte from reaching the anode. It could
be that like in the lead acid battery it doesn't play any role in the chemistry,
it's just a physical barrier to keep the anode and cathode materials from
touching (shorting).
I did some Googling ("lithium ion battery separator") to see if I could figure out what the separator does. Was surprised when I got a lot of hits (386,000 hits!) on this obscure subject. There are actually Google images of a lithium ion separator, but that doesn't help. First active link is a company Celgard that specializes in making lithium ion separators. We learn they are made of polypropylene (about 20 um thick), but their description of its purpose leaves something to be desired:
"They provide a barrier between the anode and cathode while enabling the exchange of lithium ions from one side of the battery to the other."At another separator company (Advanced Membrane Systems) I found actual separator data sheets. They have two types: shutdown and non-shutdown (whatever that means). One is "designed to reduce the effect of internal short circuits in primary and secondary lithium ion batteries". Aside from saying the porosity is 70%, there is little else. There is no mention of any ion selectivity. It's beginning to look like separators are just simple physical barriers to prevent anode to cathode shorts.
DuPont is building a plant to make a new nano-technology lithium ion battery separators for 20% of the world electric and hybrid vehicles. Here is what they say:
"Within each battery, the separator is a sheet positioned between the two electrodes. It functions as a barrier that prevents the electrodes from touching and shorting while letting lithium ions pass back and forth to allow the charge and discharge of the battery."But then they make this interesting claim:
"DuPont Energain battery separators can increase (battery) power 1530%".OK, like all marketers they probably exaggerate, but translated, what they seem to be saying is that the separator is one of the limiting items in setting the maximum discharge current of the battery. It is tempting to say the impedance of the separator must therefore be a significant term in the battery's ESR, but not necessarily. The separator is only 20 microns thick, it has almost no mass, so it might be that its a hot spot (it depends on how thermally conductive the electrolyte is), and its temperature rise is the limiting item.
Lithium cell protection devices
Translation
of below: lithium ion cells usually have built-in a non-linear resistor
(called a PTC) that is in series with the output. If the PTC (not the cell)
gets too hot, it's resistance goes way up. It acts like a solid state fuse
and will limit discharge energy if the cell is shorted, but because it's
a resistor it increases the ESR and losses of the cell. If pressure rises
within the cell, the cell is designed to come apart in a controlled way
that opens the electrical circuit and provides a pressure release for the
gases. Much of the protection for a large lithium ion battery pack (like
in a car) is provided externally.
-- The PTC device built into the cell acts as a protection to inhibit high current surges; the circuit interrupt device (CID) opens the electrical path if an excessively high charge voltage raises the internal cell pressure to 10 Bar (150 psi); and the safety vent allows a controlled release of gas in the event of a rapid increase in cell pressure.Read a paper that pointed out there is a trade off in the design of high power lithium ion batteries. The energy density depends of the volume of the lithium anode, whereas the power density (maximum discharge current) requires the electrode have a large surface area. In other words if you want high current the lithium electrode is thin with a large surface area, but if you want to store a lot of energy per lb, then you want thicker lithium electrodes.In addition to the mechanical safeguards, the electronic protection circuit external to the cells opens a solid-state switch if the charge voltage of any cell reaches 4.30V. A fuse cuts the current flow if the skin temperature of the cell approaches 90°C (194°F). To prevent the battery from over-discharging, the control circuit cuts off the current path at about 2.50V/cell. In some applications, the higher inherent safety of the spinel (manganese) system permits the exclusion of the electric circuit. (source -- http://batteryuniversity.com/learn/article/lithium_ion_safety_concerns)
Reading papers studying intercalation of lithium into graphite initially confused me.
Wow, this is tricky. I finally figured out what papers are doing (I think) are studying LiC6 for use as the anode material in place of metallic lithium, which is used as the anode in primary (non-rechargable) batteries, but can't be used in rechargeable batteries. What they are measuring is the effect on cell voltage (via delta gibbs free energy) relative to pure lithium metal. So in the paper below where they find 88 mv exothermic for lithium diffusing into the carbon, I think this means an 88 mv reduction in cell voltage (2.78V in an old paper) if LiC6 is used in place of lithium metal. The paper summs up explicitly by saying that lithium intercalated into graphite (LiC6) can replace metallic lithium at the anode (ambiguous in a rechargeable cell!) with negligible loss of cell voltage.How much lithium?A paper describing the diffusion of lithium into graphite (eq to reduction at cathode) says it is exothermic. [G = H - TS] They measure delta H = -10.3Kj/mol and delta S = -6.5J/mol-K. This says the Gibbs free energy (negative for exothermic) from enthalpy change is about -100 mv, but the entropy change in the equation has the opposite sign, so it reduces the voltage by 20 mv with the result that lithium reducing at this type of cathode would contribute about 0.086 V to the battery voltage.delta H = 10.3 Kj/mol/96.5 Kj/mol = -0.106V
-T delta S = (6.5J/mol-K x 298K/1,000)/96.5 Kj/mol = 0.02VThey describe the open circuit voltage of the cell as the difference in the 'chemical potential' of the lithium atoms in the anode and in the cathode.
1 gram of lithium
(1/7) mole
= (1/7) x 6 x 10^23 atoms
Li+ charge
1.6 x 10^-19 coulomb
charge carried by 1 gm Li+ (1/7) x 6 x 10^23
x 1.6 x 10^-19
= 1.37 x 10^4 (13,700 coulomb)
Ah/gram of lithium
13,700 coulomb/(3,600 coulomb/sec @ 1Ahr)
= 3.8 Ah
check
Since it typically takes 3 or 4 times the minimum lithium, then a large cylindrical (18 x 65 mm) lithium ion battery with a 2.4 Ahr probably contains two to three grams of lithium. For a GM Volt 16 kwh battery (assume 3.6V cell) this works out to [(16,000 wh/3.6V)/3.8 Ah/gram = 1,170 grams or 1.17 kg of pure lithium. As the paper details, the amount of lithium needed is probably, they figure about x3.5 times higher or 4.2 kg of (pure) lithium 1per GM Volt battery pack.
Range rule of thumb
Range rule
of thumb for electric cars:
1 Kwh battery drain <=> 3 miles (range)
For GM Volt this would give 10.5 Kwh (latest usable Kwh) x 3 miles/kwh = 31.5 miles (25 to 40 miles is current estimated range for electric mode). Above rule of thumb is also very close to a 2010 Volkswagen presentation for it coming electric car.
123 Systems
The chemistry
of the cylindrical lithium ion cells made by 123 Systems, widely used for
hand held drills (etc) is lithium iron phosphate (LiFePo4).
Gm Volt and Nissan Leaf lithium ion batteries
Some viewed the
face off for GM's Volt's battery as a face off between lithium iron phosphate
(123Systems) and lithium manganese phosphate (LG Chem). GM chose a lithium
manganese phosphate (or lithium manganese oxide) battery made by LG Chem.
Nissan has apparently made a similar choice for it Leaf. Wikipedia says
the Leaf battery is lithium manganate, but elsewhere it's called lithium
manganese oxide.
GM Volt uses an active liquid based heating/cooling system for its 16 kwh (400 lb) battery to minimize lifetime degradation. In contrast the Leaf 24 kwh (660 lb) battery uses just simple fan cooling. A Tesla engineer charges that Nisson has underengineering the battery and its going to degrade badly. Leaf says it will hold 80% of its capacity after ten years, but interestingly battery kwh degradation is excluded from the Leaf warranty.
GM Volt prismatic lithium ion battery (1/4/11)
The type of
cells used in lithium ion batteries in the first electric cars (Leaf and
GM Volt) are called prismatic, which as far as I can figure out just means
they are rectangular in a plastic package (easy to stack). According to
a story on the GM Volt in Popular Science (2008) (modified by latest cell
count) the GM Volt 16 kwh battery pack consists of 288 cells wired x3 parallel
and x96 is series. For 3.6V/cell this gives a 346V battery voltage (open
circuit). Each cell is rated [16,000 kwh/288 = 15.4 Ahr x 3.6V]. Peak battery
pack power (GM video interview with battery engineers) is 110 kw, which
is [118A/cell x 3 in parallel = 354A = 110,000kw/0.9 x 346V]. With
a 10% full load sag (my guess) implies a cell ESR of [3 mohm = 0.36V/118A].
Cell summary (lots of guesswork still)
# of cells
288 (wired x3 parallel x 96 series)
Ahr
15.4 Ah @ 3.6V
Peak current 118A
Peak power
382 watts
ESR
2 to 3 mohn (3 mohm assumes 10% sag at peak current)
weight
400 grams (assumes 250 lbs of 400 lbs pack is batteries)
Above derived spec looks reasonable based on below (from my hybrid car essay). It shows a 2004 lithium ion battery spec I found on the Korean LG Chem (battery supplier to GM) web site. The GM battery cell looks like this LG Chem prototype vehicle cell of six years ago scaled up about 50% in Ahr rating and weight. Even my guessimate 3 mohm ESR looks in the ball park (with 150% scaling it could be 2 mohm). At 2 mohm it would make the power loss each cell at peak power = 118A^2 x 2 mohm = 28 watts, which is 8kw (!!) = (288 cells x 0.028 kw/cell) for the whole battery pack at peak power. From an overall efficiency point of view this is pretty good (dissipating 8 kw in battery to delivier 110 kw, or about 93% efficient @ peak power).
2004 LG Chem lithium ion (prismatic) battery spec
E1 Ah
10 Ah @ 3.6 V
Weight
245 grams
Dimension
20 cm x 9.4 cm x 0.7 cm
ESR ?
3 mohm = AC impedance @ 1 khz
Max continuous discharge 30 A
(P/cell @ 30A = 30^2 x 3 mohm = 2.7 watts)
"short circuit test Z"
50 mohm (I pk = 3.6V/ (50 + 3) mohm = 68 A)
Popular Science on GM Volt lithium ion battery (2008)
Here's a cross section
of the battery. This shows how important the separator is a prismatic lithium
ion battery. No wonder that so many companies were investing heavily in
separator technology and building new separator plants!
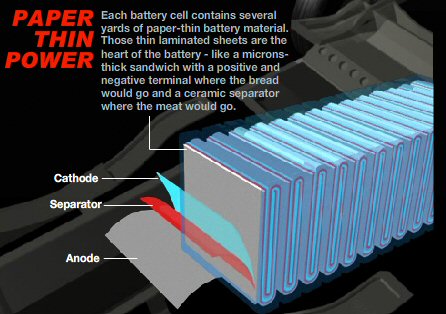 .
. 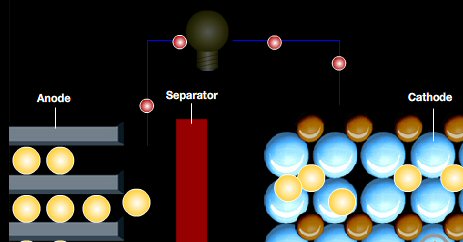
'yards' reference (if true) would imply each cell
is a huge number (dozens) of layers in parallel
(150% x 2004 LG Chem cell outside area is only 0.03
m^2)
source --- Popular science graphics on GM Volt battery
(2008)
High power batteries
MIT
Professor Sadoway's liquid metal battery
(update 9/11/14)
When I created
this essay in 20110 Prof Sadoway's concept for a liquid metal battery I
found was very interesting, but experimentally it was nothing more than
a tiny proof of concept cell running in his lab and he was seeking funds
to commercialize it. Well it's now Sept 2014, about four years later, and
I stumbled onto a reference to Sadaway's liquid metal battery in the news.
During this time a company has been formed and some product should soon
appear. The company has also had a name change, it was 'Liquid metal battery
corp', but is now 'Ambri' (derived from Cambridge) [http://www.ambri.com/].
Their web site indicates they have a staff of 38, which probably includes
some students. Total is one of their investors and a Total chemistry Phd
sits on their board. Sadaway is a consultant. Sadaway's former grad student
who co-developed with him the battery, David Bramwell, is the chief technology
officer of Ambri. News articles say they got 15 million in funding from
Total, Bill Gates and Khosla Ventures (Khosla guy also sits on the board).
In spring 2014 they announced they had raised an additional 35 million
in funding.
-- For our initial chemistry, we worked with magnesium (Mg) and antimony (Sb) electrodes and have since moved to a chemistry with a higher voltage and lower cost. (They say this and right below is a figure showing Mg and Sb electrodes! Sadaway in his 2012 TED talk also drew a Mg, Sb cell on the blackboard.) They link to a 2013 technical article 'Liquid metal batteries, Past, Present, Future', which I find discouraging. The article is behind a paywall, but the table of contests shows five different elements under 'future work', which I interpret to mean they are still fooling about with basic chemistry. A recent tech article (Aug 2014) about the battery says they have "more or less replaced the antimony with a much cheaper metal", without identifying the metal!
Product (9/14)
Sadaway in
his talks shows huge aluminum cells and speaks of this as the goal, but
the first (and only?) product to come out of Ambi so far is tiny
55 Wh cell (one sketch shows a box only 4" on a side!) 36 of these are
stacked up to for a 2 KWh module and ten of these for a 20 Kwh module.
A planned later cell (beta core) looks like it might be a 100 Wh. (Other
documents point to a 200-250 Wh cell) When stacked up, this will be 35
Kwh storage with a 17.5 Kw rating. This probably means their basic 55 Wh
cell is something like (0.5v x 27A x 4 hr). One of these shipping container
sized systems with 500 Kw (2 Mwh) rating would require about 10,000,
200 wh cells!! (Yikes, this is far, far, far from the huge aluminum tubs
Sadaway suggests is the goal.)
There is no cell spec, no Voc, no hard material info. And no deliveries yet, one prototype delivery is scheduled for end of 2014. The factory goes into operation in 2015. If enough of the these modules were stacked up to fill a shipping container, it would have a 2 Mwhr rating, and they show it mated with a large cabinet of power electronics to mate it to the grid. It's clear nothing has firmed up, as every reference has different specs for the basic cell.
A problem always passed over is that these cells cannot sit quietly waiting for demand. Current needs to be kept flowing through them to keep them hot and the metals molten.
On the plus side tests of 1,000 cells have demonstrated long cycle life. Efficiency for for four hour charge and four hour discharge is 80%, which I guess means 10% loss each way for current equivalent to peak power rating of 1/4th the kwh rating. While the company is (or was) based in Cambridge they have built a 4,500 Sqft factory in Marlborough to manufacture cells.
----------------------------------------------------------------------------------------------------------------
I wrote most
of the liquid metal battery section when I was just starting to learn about
batteries. Here is a much later overview. The anode in this battery (top)
works pretty much like the zinc family with its magnesium anode oxidizing
to form positive metal ions (mg2+). (Here, unlike with zinc, there is no
entropy increase with a atoms going into soluution from a crystal, because
the metal magnesium is already a molten liquid.) WebElements shows this
reaction to be strongly exothermic (Eo=2.4V), but this is likely for solid
magnesium ionizing.
At the cathode antimoney looks like a lusterous metal, but it is classified as a mellaloid, and when it is reduced it acts like a non metal and releases into solution negative ions (Sb3-) . I cannot find Eo for this reaction, but I suspect it's not exothermic (It's not in WebElements). These two ions combine to form magnesium antimonide, which I assume is sort of an ionic molecule. Sadoway's patent gives the disassociation energy of magnesium antimonide as 0.42V, so presumably it releases 0.42V when it forms. So as the battery runs both the liquid magnesium anode and liquid antimoney cathode shrink as they release ions which (partiall?) join up to form (ionic?) molecules of magnesium antimonide.
Cell charging
looks like its an electrochemistry cell. My working assumption is that
while the Mg2+ and Sb3- may tend to cluster together Mg3Sb2 = (3Mg2 + 2Sb3-)
there are still free Mg2+ and Sb3- ions, so when current is runs through
the cell, they are separated and both are driven back to their electrodes
where they plate out.
anode
Mg => Mg2+ + 2e-
Eo=+2.4V ? (from WebElements)
cathode
Sb + 3e- => Sb3-
Eo=?
With such a
large voltage from magnesium ionizing it would seem to indicate that the
cell voltage would probably be pretty high (2.4V ? anode + 0.4V electrolyte
forms ? +/- cathode reduction), but the patent indicates the cell
voltage is more like 1 V.
------------------------------------
All liquid:
two liquid metal electrodes and a liquid electrolyte. Self assembling!
The density (& metal surface tension) of the three layers keeps them
separate automatically. Top electrode is metal magnesium (#12) and the
bottom electrode is metalloid antimony (#51). Both electrodes dissolve
in the electrolyte, the metal magnesium losing an electron (forming a positive
ion), and the metalloid antimony acting as a non-metal gaining an electron
(forming a negative ion). In the electrolyte the two ions attract forming
magnesium antimonide. Sadoway says his approach to a high current battery
was to start with the highest current cell he knew about, an aluminum cell
that can run at 500kA, and ask if it could be modifed to store charge and
run reversibly.

Prof Sadoway's MIT World lecture 6/5/2010
(screen capture)
(Sadoway video) Antimony by itself is a metal. It's lusterous, ducticle, conducts electricity. But put antimony in the presence of a "really good metal", i.e. a really strong electron donor, like magnesium, and antimony begins to act like a non-metal and accepts electrons even though it's an electron conductor. "It's due to the difference in electronegativity."
magnesium (#12)
1.31 electronegativity
metal
44 nohm-m
antimony (#51)
2.05 electronegativity
metalloid 417 nohm-m
tellurium (#52)
2.10 electronegativity
metalloid
carbon (#6)
2.55 electronegativity
non-metal
Mg (liquid) => Mg2+ + 2e-
magnesium dissolves and liberates electrons
Sb (liquid) + 3e- => Sb3-
antimony dissolves and consumes electrons
On battery discharge magnesium atoms release electrons to the terminal (-terminal) and float off in the electroyte as positive ions, while at the other terminal (+terminal) antimony atoms accept electrons coming in the terminal and float off into the electrolyte as negative ions. So the current inside the cell is a combination of positive (Mg+) ions and negative (Sb-) ions drifting in opposite directions. (Not sure what to make of the 3 to 2 ratio in the equations, but it might mean the liquid magnesium accumulates x1.5 times faster than liquid antimony.) In the video he implies during lab test the molten electroyte salt was they used zinc cloride. He said when they doped it with tellurium, a metolloid like antimony, telluirum metal accumulated at one terminal.
Reversible ambipolar electrolysis
Sadoway in his lecture
jokes that electrolysis is putting current through "dirt" to make metals
like aluminum, magnesium, titanium. Technically Sadoway calls the liquid
metal battery 'reversible ambipolar electrolysis', so Sadoway sees this
as a special case of electrolysis, making metal. Note the figure below
shows when the cell is charged liquid metals are generated at both
electrodes.
Reversible ambipolar electrolysisIn the video he makes the point that interfaces between both electrode and electrolyte, unlike in most batteries, moves depending on the charge of the battery. Why this is big deal? He explains, in a lithium ion battery the metal lithium is embedded in a carbon matrix. In order for the lithium ions to 'dissolve' into the electrolyte they must first solid-state diffuse through the carbon matrix to reach the electrolyte, and he says the lithium ion battery designers are always struggling to improve the diffusion. His battery has no "solid-state diffusion" problem, it has better "kinetics". The implication I think is that is that his almost zero diffusion path makes the battery ESR (battery internal resistance) very low. ESR must be very low if a cell to efficiently handle huge currents (500,000A was mentioned).
Electrolysis is usually using the power of DC current to pop oxygen off a metal oxide in solution with the resultant ions separating and metal depositing at one electode and gas (oxygen or Co2) released at the other electrode. When the liquid metal battery is being charged, electrolysis is happening because the current tears apart the (two metal) 'reaction compound' (as called in Sadoway's patent) in the electrolyte, and liquid metals deposit at both an electrodes, hence 'ambipolar'. The separated liquid metals remain in the cell [in an aluminum cell the aluminum is tapped off] and chemically want to react (rejoin) in the electroyte, so the cell is reversible.
First is Sadoway
talking about the battery in his office and second is a nearly hour lecture
to what look like MIT parents.
http://www.technologyreview.com/video/?vid=264
http://mitworld.mit.edu/video/800
Paper on liquid metal batteries (2010, German)
http://arxiv.org/pdf/1005.5046.pdf$$yPreprint
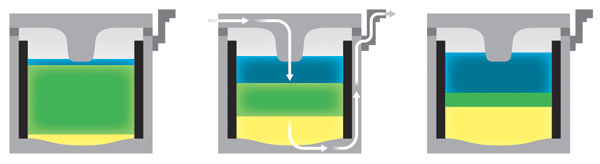
discharged
charging
charged
(poorly drawn -- arrow represents electron flow!)
(top) blue, magnesium, negative battery termnal
green, (mostly) magnesium-antimonide + (little)
molten salt electrolyte
(bot) yellow, antimony, positive battery termnal
During discharge: magnesium (blue) is the anode and
antimony (yellow) is the cathode
magnesium dissolves forming plus ions
antimony reduces to negative ions
source: Technology Review (2009)
A discharged battery has most of the magnesium and antimony atoms in solution ions (joined as magnesium antimonide). During charging electrons flow from the magnesium electrode (top, blue) into solution neutralizing positive magnesium ions into pure metal atoms, which then join the magnesium electrode causing it to grow. At the same time electrons flow from solution to the antimony electrode (bot, yel) neutralizing negative antimony ions into pure metal atoms, which then join the antimony electrode causing it to grow.
As I think about it, here is what I figure must happen at the ion level. The (downward) electron current flow through the cell at (Io) must within the electrolyte be carried by two (parallel) ion flows each at (Io/2): positive (Io/2) magnesium ions flowing upward, and negative (Io/2) antimony ions flowing downward. In each case the ions are repelled by the voltage of the electrode where they are created and attracted by the voltage of the opposite electrode. How does this work at the electrode/electrolyte interfaces?
Top electrode/electrolyte interface-- As the cell charges magnesium and antimony come out of solution, so the metal electrodes grow and the electrolyte size shrinks. During discharge, the process is reversed, the metal atoms of the electrodes become ions again, shrinking the electrodes and growing the electrolyte size. Tech Review says this is an unusual property for batteries.
Take the top interface. It must mean that half the electrons coming off the top electrode break apart a magnesium antimonide pair (molecule) adjacent to the top interface by adding an electron to the positive magnesium ion of the pair neutralizing it to a pure metal where it joins the electrode. This frees the negative antimony ion, which now diffuses downward to the bottom electrode as part of an Io/2 (antimony) negative ion current flow. The other half of the electrons coming off the top electrode must meet and neutralize an (Io/2) flow of positive magnesium ions that have diffused up from the bottom electrode/electrolyte interface.Bot electrode/electrolyte interface
Obviously at the bottom electrode/electrolyte interface the same basic thing (in reverse) must be happening. Half the electrons entering the bottom electrode must come from breaking apart a magnesium antimonide pair (molecule) adjacent to the interface pulling the electron off the negative antimony ion of the pair neutralizing it to pure metal where it joins the electrode. This frees the positive magnesium ion, which now diffuses upward to the top electrode, as part of an Io/2 (magnesium) positive ion current flow. The other half of the electrons entering the bottom electrode must come from an (Io/2) flow of negative antimony ions that have diffused down from the top and have been neutralized at the bottom electrode/electrolyte interface by release of their electron.
-- The two ions in solution form (dissolved) magnesium antimonide. During charging the current decomposes this reaction product (magnesium antimonide) and the ions migrate to the electrolyte/metal interfaces where they are neutralized and join the liquid metal at the electrodes. The cell needs to run at 700C to keeps the metals molten, but this is not so hard when the cells are huge and can use electrical losss to self heat. A battery the size of a shipping container could deliver 1 Mw for several hours. In the video he says large cells can hold their temperature for days.Sadoway liquid battery patent application
Publication Number: US20080044725
Search applications
Inventor name
Sadoway
Inventor town
Watertown
Another reference is the 2006 Master thesis of Sadoways grad student, David Bradwell, "Technical and economic feasiblity of a high temperature self assembling battery.
(some patent excerpts)
-- charging
current drives electrolysis of the cell electrolyte
-- products
of electrolysis remain within the cell available to react and recombine,
allowing it to discharged on demand and to provide external power.
-- anodically
deposited liquid metals can serve as an electrode in a cell
-- anode candidates
are the metalloids: antimony, tellurium and bismouth
-- these metalloids
as non-metals in forming compounds with metals, but in liquid phase they
behave as liquid metals in that they are electronic conductors.
-- This confluence
of properties makes possible the extraction of a liquid phase electronic
conductor from a molten salt by oxidation as part of ambipolar electrochemical
metal extraction in which metal is also extracted by reduction at the electrode
of opposite polarity.
-- Metalloid
electrode is positive (yup, on discharge it accepts electrons, which means
positive current is going out. It is the positive terminal of the battery.)
(In effect)
Open
circuit charged cell. Both metals would 'like' to disolve, but neither
can convert to ions (apparently only ions can dissolve) as there is no
where for the Mg to throw an electron (to make Mg+) and nowhere for Sb
to get an electron (to make Sb-)
My sketch
of the cell shows there must be a (linear) gradient of both Mg+ and Sb-
across the cell. Current is the sum of a linear up and linear down slope.
At the top Mg/electrolyte boundary Mg+ flood across into solution (max
of gradient) and at the bot (Sb/electrolyte) boundary Sb- flood across
into solution.
-- Preferred
metals for negative electrode: magnesium, potassium, calcium, lithium,
sodium (alkali and alkaline-earth metals) and cadmium, zinc.
-- Preferred
metalloids for positive terminals: antimony, bismouth, arsenic, and tellurium,
selenium
-- known electrometallurgical
cells generate gas at the anode
-- reaction
rates at the boundaries have the highest reaction rates known in electrochemistry.
-- expected
current density id several amps/cm^2 (riviling aluminum smelter) with efficiency
of 80% (what does this mean?)
-- one self
assembling chemistry is Mg3Sb2 and operates at 700C
-- DC to DC
efficiency 80%, discharge time 4 to 10 hr, 10kw to > 10 Mw
-- candadates
for electrolyte in magnesium-antimony cell are sodium sulfide, calcium
sulfide, lithium sulfide, magnesiuum sulfide, potassium sulfide
-- magnesium
antimony cell consists of self assembling immiscible liquids, operating
at 700C
top:magnesium (1.5 g/ml, melts 650C),
middle: appropriate electrolyte containing magnesium-antimonide (4
g/ml, melts 1,245C)
bot: antimony (6.5 g/ml, melts 630C)
-- to limit
the electrolsis products to the liquid elements of the electrodes the electrolyte
will have 'free energy of formation' 'more negative' than that of the 'reaction
compound'
-- an additive
to an electrode (like 10% mole lead to antimony) will cause the cell open
circuit voltage to drop as the antimony dissolves and the lead concentrates.
This provides a signal that the cell should not be further discharged.
-- before
charging, most of the magnesium and antimony are present as ions dissolved
in the electrolyte
-- (at magnesium/electrolyte
boundary during charging) electrons are unable to travel into the electrolyte
because it is electronically insulating, so electrons combine with magnesium
cations (positive ions) at the interface creating metallic magnesium which
accrues to the negative (magnesium) electrode.
-- current
is carried across the electrolyte by Mg2+ (moving upward) and Sb3- (moving
downward).
(Sb3- is a 'trivalent antimony ion') (mg2+ is a 'divalent magnesium cation')
-- to keep
some metal at the negative electrode at the end of discharge an excess
of the negative metal is provided relative to the 'stoichiometric ratio
of negative and positive electrodes corresponding to the reaction compound
(Sb:Mg:2:3)
-- graphite
is suitable for cell walls as it is 'resistant to attact' by the molten
salt electrode
-- side by
side system could be composed of metal zinc (7.1g/ml), liquid metalloid
tellurium (6.3 g/ml), with reaction product zinc-tellurde dissolved in
zinc cloride (2.9 g/ml). This molten salt will dissolve more than 20 mole
percent of the reaction compound (ZnTe). It provides conductivity by means
of 'zinc cations (Zn2+)' and 'chloride anions (Cl-)'. (What? Is this an
error? Doesn't this evolve clorine gas! Later it says zinc and tellurium
ions get deposited at the electrodes)
-- In general
cell voltages are small. The 'disassociation potential' of magnesium antimonide
is 0.42V. The actual cell voltage is influence by the actions of the ions
in the electrode, as expressed by the Nernst equation. The activities likely
exhibit large nonidealities which may shift the 'open circut' voltage to
more than 1 volt. (not clear). (later an array of 96 cells in series is
described as having an 'open circuit' voltage on the order of 100V) (there
is no other v-i curve and also no discussion of chemical pootentials )
German paper
-- Electrolyte
is a molen salt chosen so it is immiscible with both the metal and metalloid,
so their ions can diffuse. It is also electrically insulating and ionizes
the reaction product. (After saying it is insulating, the paper then says
the electrolyte needs high conductivity to keep ohmic losses low!)
The metal and melloid have to be highly soluable in the electrolyte.
-- 30 Mw of sodium sulfur batteries are in use now to stabilze grid power in Japan
My thoughts
This seems
like a fundamentally new idea for a battery. Pour in three (very hot) liquids
into a big tub with electrodes. The liquids self-separate due to density
differences forming three horizontal layers. Presto/chango you have got
a super high current rechargeable battery! And it could solve a real
need in the energy world by providing energy storage for the smoothing
of wind and solar power. Of course, Sadoway is selling this concept and
the devil is in the details, but this liquid metal battery looks like it
could be a fundamental breakthrough in building really big affordable batteries.
The electrolyte volume changes enormously. When the battery is discharged, the (liquid) electrolyte must be mostly Mg+ and Sb- ions! When delivering power, the metal magnesium (element 12) electrode gives up electrons to go into solution (forming mg+), and the antimony (element 51) electrode (on the periodic chart a metaloid or semiconductor) must here be accepting electrons to go into solution (forming sb-).
From an entropy point of view charged/discharged states make sense. Entropy increases as a system of isolated atoms evolves to a system of mixed atoms. A run down discharged battery has high entropy with a large volume of mixed ions. Charging the battery organizes the metal atoms is a highly ordered state (the Mg and Sb atoms are separated) decreasing the entropy.
[Entropy is equal to the log of the number of states a system can take. There is only one way a deck of cards can be ordered, so an ordered deck of cards has low entropy. There are many ways a deck of cards can be disordered (by shuffling), so a shuffled deck of cards has high entropy.]Don Sadoway's Wiki article
Sodium
sulfur high power battery (Japan)
While Sadoway's
liquid metal rechargeable battery is still at the lab stage, the Japanese
have designed a high power (12 Mwh) molten (350C) sodium sulfur rechargeable
battery for grid smoothing that has reached the stage of commercial production.
One reference says Ford invented this battery in 1966, and I know they
played around with a high temperature battery for years, which I always
thought was a joke for cars. And it is!
160 Mw have been installed in Japan to smooth solar and wind power, and 4 Mw from the same Japanese company were recently installed in a Texas town to ride out black outs. They rate its lifetime as 15 years. Sodium sulfur has a long history as this is the battery Ford played around with for about 20 years (1970 to 1990), and since the mid 1990 a few Mw prototypes have been in use by utilities.
The battery has an anode of molten sodium, a cathode of molten sulfur and between them a ceramic separator, variously called a 'solid electrolyte' and a 'beta alumina of sodium ion conductive ceramic'. Sodium is a metal so it conducts well, but non-metal sulfur is poor conductor so is embedded in what Wikipedia calls a "carbon sponge". Each cell is sealed in a corrosion resistant steel container. It's chemistry looks very simple. During discharge the sodium ionizes to Na+ ions, which pass through the ceramic separator and join up with the sulfur to form sodium polysulfide (Na2S4). The sodium metal anode slowly shrinks and the cathode sulfur slowly converts to sodium polysulfide. The sodium ions carry all the current. When charging the process is reversed, sodium polysulfide is oxidized to neutral sulfur and Na+ ions. The Na+ ions are driven back through the ceramic separator rebuilding the quantity of sodium metal at the anode. Here's an NGK diagram:
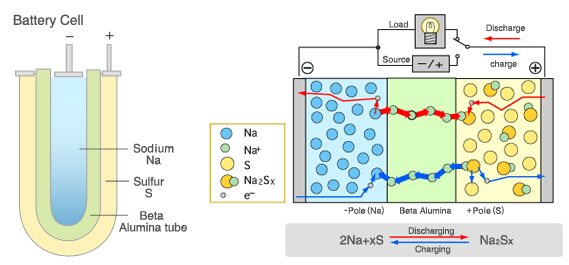 .
. 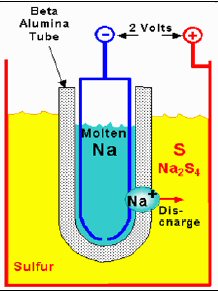
Molten sodium-sulfur high power rechargeable battery
Big red/blue arrows show the sodium ion movement across
the separator,
and small red/blue arrows show electron movement.
(sources -- http://www.ngk.co.jp/english/products/power/nas/principle/index.html)
http://www.ettexas.com/projects/docs/NaS_Battery_Overview.pdf
Chemistry
The above sketch
shows the cell voltage at 2V (textbook says 2.1V), but Wikipedia ('Molten
salt battery') says the oxidation potential of sodium is 2.71V. Seems inconsistent
as I have never seen a battery yet (except Volt's first cell) where the
cathode didn't contribute some energy to the output, but I suppose it's
possible that the formation of Na2S4 at the cathode does take 0.61 ev of
energy.
anode
Na => Na+ + e-
Eo = 2.71V
cathode
S + 2e- => S2-
(Na+ and S2- form sodium polysulfides)
textbook 2Na +3S => Na2S3
Two lithium (or two sodium) per sulfur atom. The delta G is described as the 'energy of formation' (Positive delta G is energy in, so negative values indicate exothermic reactions.)
2Na + S => Na2S
delta G = -350 kj/mol (x 0.0104 ev/(kj/mol) = 3.63 eV)
2Li + S => Li2S
delta G = -425 kj/mol (4.40 eV)
------------------------------
Separator losses
Found a good
sodium sulfur paper from a US company (Trans Ionics) working on an improved
version of the sodium sulfur battery. The paper has the first data I have
seen on a separator (here a solid aluminum oxide that only passes lithium
ions.). They give Ford data that shows the separator looks like a resistor!
It has an ohms/m^2 spec, linear with thickness, and they give voltage drop
data:
Voltage drop (of separator) @ 7.5 kA/m^2 = 304 mv
@ 350C (1 thickness)
= 24.8V @ 25C
(1 thickness)
As this data shows the resistance beta (Al2O3) separator is wildly temperature sensitive. It drops hugely with temperature, another reason to run the battery really hot. The implication is that this 300 mv voltage drop is typical and was a problem with the Ford type cells, but elsewhere they say Ford demonstrated high cycling efficiency of around 90% indicating it was not a problem. The plan to drastically reduce the separator thickness (thin film separator), which leads to a huge resistance reduction and voltage down to 2 mv.
In fact this company says the high resistance of the separator and electrolyte has reduced the sodium sulfur energy density in the cells of the commercial Japanese battery by a factor of 5, and the structural and thermal overhead which reduces the density for the battery pack by another factor of 2. In other words sodium sulfur theoretically is capable of 1.25 kwh/kg (eq to 376 kwh (!) for a Leaf type 300 kg battery) whereas the commerical battery pack is achieving 0.12 kwh/kg. This company is claiming that their thin film separator will reduce losses so much that their advanced sodium sulfur battery will be able to operate at 25C to 100C. (They admit a little problem is making a 5 micron separator that is pinhole free! It will be plated onto a microporus material for strength.)
What?US thin film company working on advanced sodium sulfur battery (/Small company, 72,000 sq ft and 3 million in government research grants over ten years.)
Sodium's melting point is 97C, so a (pure) liquid sodium battery has to be at least this temperature. Transionics is planning to use some (unnamed) sodium compound to operate below 97C.Some loss in the separator of a liquid sodium battery is OK, as it functions as an internal heat source to keep the cell up to temperature.
The beta ceramic seperator in liquid sodium batteries is more than a separtor. It's part of the structure of the battery, it needs physical strength as it is shaped into a tube to hold the liquid sodium.
http://www.transionics.com/NaS_battery.pdf
------------------------------
The Wikipedia
provides no info on the chemistry of the sodium sulfur battery, but this
is a related battery (lithium sulfur), which has not made it out the lab,
and its chemistry is described. At the cathodes a series of reductions
of sulfur occur, where sulfur changes to lithium (poly) sulfides (as you
go right below each sulfur has merged with more Li+ ions requiring the
acceptance of electrons.
S8 => Li2S8 => Li2S6 => Li2S4 => Li2S3
Apparently a common problem to rechargable batteries like this is that when they are charged and metal anode reforms dentrites slowly grow. This can lead to puncture of the separator and shorting the cell.
Wikipedia says the above reaction is "analogous to those in the sodium-sulfur battery". Sulfur is not a good conductor so to get conductivity the sulfur is mixed ('coated') with carbon with work focusing on high tech carbon nanofibers, probably indicating this is real engineering problem in these type of batteries. Wikipedia says each sulfur atom in a lithium sulfur battery can 'host' two lithium ions (this does not show in the equation!), whereas in lithium ion battery cathodes each host atom accepts 0.5-0.7 Li+, so the absorption of lithium ions increases the energy density of the battery.
Commercial use
The world's only
manufacturer of the sodium sulfur battery is NGK Insulators of Japan, and
it looks like there are only a handful of installations. Their 12
Mwh (2 Mw for 6 hr) battery is a stack of 40 hermetically sealed units
each rated at 50 kw. A cutaway view shows a 50 kw module is made up of
maybe 100 to 150 (sealed) cells, so my guess is all 125 cells or so are
in series making the output 250V @ 200A [50 kw = 200A x 125 cells x 2V/cell].
The units are stacked up in an array about 15 feet high by 25 ft long with
each module weighing about 4 tons. If there 125 cells/module, then the
weight/cell is appox [64lb = 8,000 lbs/125 cells] and each 64 lb (2V) cell
has an Ahr rating of [1,200A = 200A x 6 hr]. The 2Mw 'battery' is probably
made up of 5,000 of these 64 lb cells [5,000 = 125 cells/module x 40 modules].
The associated 2 Mw inverter occupies a stack of 11 six foot high cabinets
extending about 25 feet. Below is a picture of the a 34 Mw assembly used
to smooth the output of a 51 Mw wind farm in Japan, the world's largest
sodium-sulfur battery installation.

34 Mw (@ 6 hr) NGK sodium sulfur battery installation
used to smooth 51 Mw wind farm
(each white building is a 2 Mw battery)
Sodium
nickel-chloride medium power battery (update 5/2013)
The press
is announcing that three (of 86) new 2.5 Mw wind turbines to be
installed in Texas windfarm by GE will be "brilliant" because each will
have advanced software and be connected to a GE durathon (sodium nickel-chloride)
battery for load equalizing. Almost none of the stories give the capacity
of the battery, which is a big tip off ! Sullivan's blog said the battery
was rated 50 kw. This is only 2% of the peak power capacity of the wind
turbine which sort of looks like a joke compared to the 66% capacity of
the Japanese installation above, but it's a start. Clearly the claimed
15-25% improvements in the wind turbine power output must come from mostly
from the software (assisted by an unknown extent by low level power absorption
capabilities of the small battery). GE has invested 100 million dollars
to build a huge (four football field long) high power battery plant in
New York, and it will supply the batteries for the new Texas GE wind turbines.
GE president touring the plant when it opened in summer 2012 predicted
it would be a 500 million dollar business in four years. 60% of the plant
is devoted to making the ceramic tubes that form the battery.
Turns out at this this GE battery is not a high power battery like the NGK battery (above). It's smaller, suitcase size, a medium power 15 kwh, 2.5 kw battery built from foot long, 2.58V, 100 watt cells in series. 21 hermetically sealed cells are packaged into a 264 lb, thermally isolated box (2ft x 2 ft x 1 ft). And it was not designed with wind turbines in mind, but to support 1-5 kw cell tower loads for a few hours. Curves on the data sheet show a 1V drop (52.5V to 51.5V) with load increase of 20A (1.6 kw to 2.6 kw) for an ESR of 50 mohm. At 2 kw load (50V x 40A) the internal ESR loss is 80 watt and 20 watt at 1 kw. These are good number only 4% power loss each way up near rated power. And since this is a molten salt battery that operates at 300C, this power is not really wasted, it's needed to keep the battery hot.
HistoryA GE white paper says "sodium metal halide" batteries were conceived in the 1970s as a solid metal, sodium chloride positive electrode separated from a liquid sodium negative electrode by a ceramic beta-alumina solid electrode tube. The open circuit voltage of the cell when the metal is nickel (another variant is iron) is given as 2.58 V, operating temperature is 300C, and energy density is 120 Wh/kg (at battery pack level). (Agrees nicely with Durathon spec sheet: 15 kwh/120 kg = 125 Wh/kg). With active cycling < 1 kwh/day is needed to maintain the battery operating temperature.
The idea for this type of molten sodium nickel chloride battery has been around for 30 years. It was invented in South Africa in 1980's as the Zebra battery, and they later sold the technology to a Swiss company (MES-DEA), whose press reports said they had invested 60 million dollars in a factory to build it. But apparently neither of these companies was able to make any money out of it, so a few years ago the Swiss company sold it to GE, one poster characterized this as GE rescuing the technology.GE thinks there's a market for a long life, rugged, medium power battery in cell phone towers, a huge business. Off grid cell towers are now powered by diesel generators, which require truck visits to refuel. GE argues that mating a battery with the diesel generator can cut it fuel use as much as 50% (GE brochure shows 39%), half the truck visits. The saving comes from running the diesel generator at its most efficient load point for just a few hours. The battery also makes sense where a grid connection to the tower is not reliable. GE has invested 100 million to convert an old turbine factory of theirs in NY to make this battery (450 jobs if full capacity reached), and there is talk they are prepared to invest 70 million more. They have a 63 million dollar order for 3,500 African, Indian and Asian cell tower batteries. 60% of the plant is devoted to making ceramic tubes for the battery. Apparently the plant is starting off with only one model, a 15 kwh suitcase size assembly of 21 cells in series forming a 50V, 50A battery.
Cell tower economics
World wide there 640k cell phone towers (of five million total) not on grid and mostly of which run on diesel power. 75k new off grid towers were projected to be added in 2012. India's telecom industry in 2011 used 3.2 billion liters of diesel fuel which cost 80 cents/liter, or 2.56 billion dollars in diesel. This makes the 63 million investment in GE cell tower batteries look pretty small. The Scientific American article goes on to say the annual cost to run a diesel generator for a base station is 14.5k. The GE contract numbers (63 million for 3,500 towers) would indicate battery cost run about 18k/tower. So if fuel saving of 50% could be obtained this is a 2.5 year payback (of 5 yr payback for 25% savings.India is mandating that much of its off grid tower transition to solar in the next few years, but solar needs a battery too, so GE will still have a market for its batteries, probably a bigger market, since the batteries need to provided at least 18 hr of power. Power usage of a rural cell phone tower run 1-5 kw with one source saying many run at 5 kw. For perspective the rating of GE's only battery is 15 kwh, but only about half of that range is really usable, so at 5 kw that translates into 1.5 hr, which would mean 12 battery boxes (min) would be needed for a solar 5 kw tower.
Solar cell towers?
Here's a picture of the first fully solar powered T-Mobile cell tower (in Pennsylvania) installed 2010. It has about 12 m^2 of solar cells, which means the max power output (bright sunlight) is 1.8 kw = [0.15 eff x 12,000 watts incoming solar power ]. Even in the desert this would be a daily average of 1/4th max or only 450 watts = (6hr/24 hr x 1.8 kw) and further north maybe half this (225 watts). This is a long, long way from 5 kw. I conclude from this that only low power cell towers can run 100% on solar, or it will take huge solar arrays to power them. (Information always missing from the PR in the press.)
Below is GE's sketch of the chemistry of its Na-Cl2 battery. This battery is closely related to the NGK sodium sulfur battery. Both have a tub of molten sodium as the anode and solid 'beta' alumina separator that passes sodium ions. The difference is at the cathode where GE replaces the sulfur (solution) with a nickel (solution). When charged, all the sodium is on one side and the other side is a nickel chloride in a nickel; aluminum chloride (NiAlCl4) electrolyte plus a nickel terminal. As the battery discharges, the sodium ions migrate across the separator and replace the nickel in solution to form sodium chloride in solution (dissolved salt) with the nickel plating out (or building up around) on the nickel electrode.
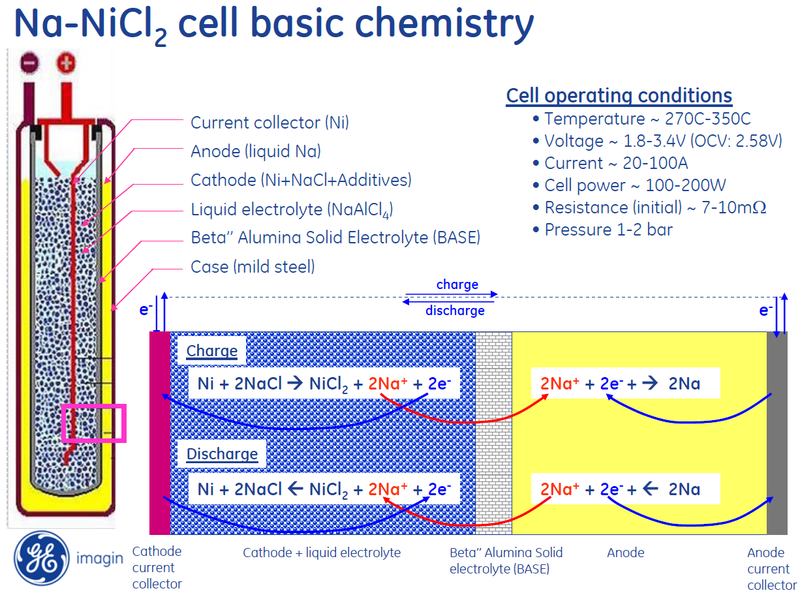
Separator passes only Na+ ions.
Discharge: sodium moves from its anode tub to form
sodium chloride (salt) in solution in cathode tub
replacing nickel which (probably) plates out on cathode
electrode.
Charge: sodium is driven back to its anode tub as
nickel from cathode electrode
goes into solution and (loosely) bonds with chloride
Max power: 300W (internal) =200W out (100A x 2V) +
100W separator losses (100A x 10 mohm (1V) x 100A)
(15,000 wh battery curves show the ESR of each of
its 21 cells is more like 2.5 mohm)
How it works
Sodium as
a group 1 alkali metal will happily (if allowed) dump its single 's' shell
electron, throwing the electron off with an energy of 2.71 eV as the sodium
atom drops in energy to become a sodium ion (na+). In electrical model
terms as the sodium atoms in the anode tub dump their outside ('extra')
electrons out the negative terminal where a +2.71V battery (relative
to the negative terminal) forms. This voltage then drives the newly created
sodium ions (Na+) across the separator into the cathode tub, which has
a (loosely bound?) nickel chloride in solution and a metal nickel terminal.
(Wikipedia says nickel chloride does not dissolve, so it apparently forms
some sort of spongy matrix around the nickel terminal.)
The electrons, having dropped their energy externally into a load, return and flow into the positive battery terminal. The electrons available at the positive terminal allow the diffusing in Na+ ions hitting it to neutralize and grab a chloride from the nickel and go into solution as an ionic compound, sodium chloride (salt). The replaced nickel in simple terms 'plates out' on the electrode, but what this really means (as a figure shows) is probably it forms an open nickel matrix around the solid nickel terminal. The voltage of the sodium oxidation is shown as 2.71V, but the open circuit voltage of this battery is 2.58V, so this would argue that sodium replacing the nickel (in the chloride) forms a battery at the positive terminals that reduces the battery voltage by 0.13V.
Separator
One of the
keys to molten sulfur batteries is 'beta-alumina solid electrolyte' separator.
It is an aluminum oxide ceramic whose crystal structure allows relatively
free passage of ions (like na+) while blocking non ions. Wikipedia says
it was invented by Ford in the 70s and 80s when they worked on the sodium
sulfur battery. For use sodium sulfur and sodium nickel chloride batteries
it is formed into thin 1.25 mm tubes using low cost ceramic manufacturing
techniques. Electrically it's modeled as a resistor. One interesting feature
is that its resistance is a wild function of temperature, it goes down
as it gets hot. It's resistance at 20C is x18 (!) higher than its resistance
at its usual operating temperature of 300C.
SidelightsDurathon specs
I read molten sodium batteries normally fail (cracking of separator?) to a low resistance (comparable to a normal cell), so a battery is still usable with a few dead cells. When the molten sulfur nickel battery was studied for use in cars, in a crash it was assumed the separator would break. I read this could take away the risk of (free) sodium, which reacts with water, because the battery can be sized so the chloride will tie up all the sodium (into salt).
energy storage
15 kwh (6 to 12 hr @ rated power)
rated discharge power
2.6 kw (1.2 to 2.6 kw)
weight (with thermal subsystem)
264 lb (120 kg)
size
25 in x 20 in x 12 in
open circuit voltage
54.2 V (21 cells x 2.58 V/cell)
discharge current (nom)
50A (50A x 52V = 2.6 kw)
charge current (max)
100A (100A x 50V = 5 kw or C/3)
ESR (typ from curves)
50 mohm (1V sag @ 20A (1 kw) additional)
Cost
?? (GE contract works out to 18k/tower)
http://geenergystorage.com/telecom/technical-specifications
This shows GE's projected savings in diesel fuel a battery can provide in cell tower usage. It shows the battery charging for 2 hr and then discharging for 3 hr, consistent with a 2.5 kw load (@ 50% usage). The projected diesel daily fuel savings is [41 => 25 liters] or 39% (= 16/41) reduction in fuel costs. At the India price of diesel of 80 cents/liter this is a savings of 12.8 dollars/day or 4.67k/yr not counting the saving is delivery costs, which should also be down 39%.
(source -- http://geenergystorage.com/telecom/brochures)
The white paper says typical numbers would be: 4 hr charge and 8 hr discharge. This battery doesn't seem at all suitable for load leveling a large wind turbine as its power rating is quite low. An interesting feature is that if the battery is cooled down and allowed to 'freeze' it will maintain its charge indefinitely.
From Wikipedia here are the resistivity of the relevant pure elements (@ 20C). How much the resistivity of sodium changes when it is melted I don't know. Sodium melts at 97 C, just under the boiling point of water, so this is the minimum temperature of the battery.
sodium resistivity
47.7 n-ohms-m x2.8
copper
nickel resistivity
69.3 n-ohms-m x4.1 copper
copper resistivity
16.8 n-ohms-m
--------------------------------------
Battery power rating (@ 6 hr) is 2/3rd peak wind power
Working the
numbers it looks like the Japanese have installed a 2 Mw (6 hr) battery
for each 3 Mw wind turbine in the farm. If the turbine operates at 33%
capacity, then the peaks at high wind speed can be fully shaved off: 2
Mw of the 3 Mw from the turbine is used to charge the battery with the
1 Mw (ideal) average fed directly to the grid. A full charged battery can
then hold the output flat at 1 Mw (ideal) av for 12 hours of low wind speed.
There is no cost info on the NGK site, but the Presidio Tx site in 2010 installed two NGK 2 Mw sodium sulfur batteries (4 Mw total). It took 24 trucks to transport the 80 battery modules from Ca port to Texas. The 4 Mw NGK Tx battery substation cost about 25 million, or 12.5 million per 2 Mw battery. The cost per 64 lb cell (including pro-rata share of building cost and inverter costs) comes out to be 2.5k [2.5k = 12,500 thousand/5 thousand cells]. If it takes one of these batteries per 3 Mw wind turbine, it makes the cost of the battery two or three times higher than the turbine!
Comparison of high power battery chemistries
The liquid
metal battery and sodium sulfur batteries are actually quite similar. They
are both composed of two elements kept separate (by a separator or density
difference), and there is no electrolyte per se. When charged, the battery
consists mostly of pure elements in the anode and cathode. As the battery
discharges, the anode and cathode elements combine to form an ionic (?)
compound that builds up. Charging the battery breaks up the ionic compound,
the ions separate, and the pure elements at the anode and cathode build
back up.
Both high power batteries have liquid metal anodes using two metals adjacent on the periodic chart: sodium (#11, group 1) for sodium sulfur, and magnesium (#12, group 2) for liquid metal. These are low density alkali and alkaline earth metals. The anode metals oxidize to (positive) ions that drift to the cathode (well maybe not all the way in the liquid metal battery) where aided by reduction electrons they from a compound (likely an ionic compound) with the cathode non-metal (or metalloid). In sodium sulfur Na+ joins with elementary sulfur to form Na2S4, and in liquid metal Mg2+ joins with elementary antimony (or Sb2+) to form magnesium-antimonide, which is described as a salt.
The differences are these: To keep the molten sodium (anode) separate from the molten sulfur/carbon (cathode) the sodium sulfur uses a solid (ceramic) barrier that is porous to Na+ ions. The compound that is created (Na2S4) builds up in cathode chamber. In battery operation the sodium chamber (anode) empties and the sulfur in the sulfur chamber (cathode) converts to Na2S4. Charging reverses the process with Na2S4 breaking up and the Na+ ions driven back across the barrier to the anode chamber.
In the liquid metal battery the magnesium-antimonide compound that forms is less dense than the liquid antimony cathode, but more dense than the liquid magnesium anode, so it floats in a layer between them. No barrier is needed to keep the molten anode element separated from the molten cathode element, because (like oil and water) the anode liquid metal is chosen to be light, it wants to float on top. The anode metal is magnesium, which is a light alkaline earth metal that weighs about 1/6 th of lead and about 1/4 th of the cathode metaloid antimony.
Thermal batteries
There is a
type of high power battery that has been used by the milatary for over
50 years, and I had never heard of them. It is a class of batteries
with molten salt electrolytes, called thermal batteries. They were invented
by the Germans and used in the V2 rocket, and continue to power many missiles,
like the Sidewinder and Patriot, as well as powering proximity fuses
for artillary shells. While generically the sodium sulfur battery with
its molten salt electrolyte could be considered a thermal battery, the
term thermal battery is generally used to mean batteries that are stored
cold, and then are warmed up to run.
The ionic salt electrolyte only become conductive when molten at 500C or so. In missiles the molten salt is held in place by magnesium oxide using capillary action. At room temperature the electrolyte is solid and unconducting, so the battery is totally inert and can be stored for 50 years with no self discharge or deterioriation. In the V2 the battery was warmed by the heat of the engine. In modern batteries they have heating chemicals inside, which are triggered first to activate the battery.
Electric car batteries
(5/11)
A friend sent
me a news article about a professor developing a fast charging battery
and asked if this was the solution to electric car range problem. Here's
my reply.
The Ecocomist magazine had a similar story about a fast charging battery a few months ago. I think I posted to it because fast charging is mostly hype. Most of these batteries don't exist, but let's ignore that and assume they do. If the battery ESR can be reduced, it's mostly a good thing because the battery is more efficient and runs a little cooler. (However a poster did point out that a big battery with a very low ESR could be considered a bomb, and he may have a point.) In fact some pretty fast charging large lithium ion batteries, 10 min recharge (!) say their manuf, have been on the market for quite a while, but when you look at products you see much longer recharge times. Why? Comes down to basic physics and cost.So what's the solution he replied, just better batteries? I continued:1) Current/Wiring
Your 'basic' electric car has 24 kwh battery and a battery voltage in round numbers of 300VDC. To recharge this battery fully in one hour I = 24,000 watt/300V = 80A. To recharge it 1/10th time (6 min) makes the current 800A. 6 min recharge means all the wiring in the charger, battery, car wiring and its plug would have to be rated for 800A! (The standard plug adopted by all the electric cars has about a 70A rating, though Nissan has also included in the Leaf a 100A connector for 480 VAC charging)2) Power source
For a 6 min charge the power is 24kwh/0.1hr = 240kw (1/4 Mwatt). Either a charging station pulls 1/4 Mw/car in real time from the grid or it needs local storage, say the equivalent to x10 cars, or 240 kwh. If the cost of a car battery is 10k, then obviously for 100k you could assemble a 240 khw battery stack. Large sodium sulfur batteries at 2 Mwh each are in production by a Japanese company for load balancing. It takes a small building to hold a 2 Mwh battery. All this cost might (might!) make sense for 'electric gas stations', but it's not something you can afford to do at home.3) Electric gas stations
A poster to the Economist article pointed out that the concept of 'electric gas stations' may not make any sense. You can't get gasoline at home, you have to go to a gas station. But in an electric car, if you can get home, you can charge up virtually for free (at least in terms of your time). So who is going to stop at an electric gas station, only those people on trips of more than 80 miles? When you think about it, the whole idea of high cost, slow, electric charging stations may not make any economic sense. And if/when batteries get better and car range expands, electric gas stations make less and less sense.Ford focus electric (23 kwh battery, direct competitor to Nissan leaf) begins production in a few months. They are featuring a 240 VAC (1 ph I think) recharge that will take 3-4 hr. By the way I was curious as to how Ford would handle the battery derating issue. Go the Volt way with fancy temp control, or the risky/cheap Leaf way with just a fan? Answer is in, Ford will be conservative with the battery and will have liquid temp control.
The high current problem is absolute fundamental to power distribution. It's the reason why we have AC power today, why Tesla/Westinghouse AC power using transformers was able to overtake Edison DC power. Fast charging means high power, and the only way to to deliver high power without thick wires is increase the voltage.Cathodic protection of shipsBut high voltage in cars leads to two really bad problems. 1st safety --- Do you want 3,000V battery to get 6 min charging? 2nd chemistry ---- A 'standard' car battery now has about 100 @ 3V cells in series. I know enough chemistry to say it is impossible to raise the cell voltage more than a few tens of a volt, lithium ion cell voltages now are close to the theoretical maximum. So a 3,000 V battery means 1,000 smaller cells @ 3V in series. Theoretically possible I suppose, but I have never heard of anybody working toward a goal like this. And without some revolution in terms of cell construction it's going to be too expensive because it has too much stuff: every cell needs its own anode, cathode, separator, electrolyte, etc.
The only other approach anyone has thought of is to swap battery packs in/out and some companies are pursuing this. This might conceivably work for large standardized fleets, like taxi or truck fleets, but for ordinary users it has two big problems. One, it requires the car be designed with a standardize battery pack (for which no specs exist). The battery is large, heavy, a key design element of the car, so this is a long way off. And two, it brings us back, in spades, to the 'Electric gas station' problem.
So long range means better and or bigger batteries or a range extender. A cute idea from one of the early electric car pioneers was a small charging trailer (he built one) that you attached and towed for long trips! It's the Volt approach with the engine, generator and gas supply external to the car.
It turns out this is remarkably easy to do. A dissimilar metal is added, which with sea water and the ship's metal forms a battery, and the battery is shorted making current. The added metal, such as zinc, must be more active than the ship's metal, i.e. it wants to dissolve (oxidize) more strongly that the steel (or copper) ship bottom in sea water), and it is hard connect (shorted) to the ship's metal.
The zinc forms the anode, the steel (or copper) of the ship forms the cathode, and the sea water is the electrolyte. Because the battery electrodes (zinc and ship's metal) are shorted, the battery voltage from the zinc dissolving (into the sea water) makes an small E field in the sea water that drives a (positive) current into the ship's steel (or copper) bottom. References show the reaction at the ship's metal is generation of OH- ions as oxygen and incoming electrons reduce water molecules, but this has no effect on the ship's metal.
Iron rusting in water
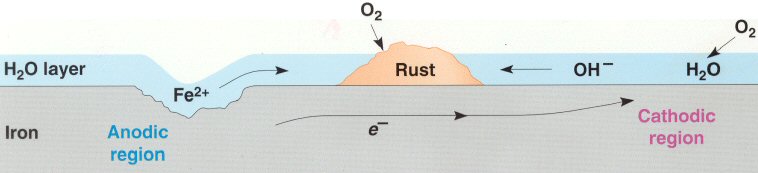
My cathodic protection sketch4Fe => 4Fe2+ + 8e-
2O2 + 4H2O + 8e- => 8OH-
--------------------------------------------
4Fe + 2O2 + 4H2O => 4Fe2+ + 8OH-
O2 + 4Fe2+ + 8OH- => 2Fe2O3 +4H2O
----------------------------------------------
4Fe + 3O2 + 4H2O => 2Fe2O3 +4H2O
The hidden trick to cathodic protection is that the created battery is shorted. The short is created (blue line in sketch) by bolting (hard connecting) the zinc to the steel. But the battery is still there, and what happens is that the 0.32V of the cell drops across the electrolyte (seawater) driving a small (ua to ma?) continuous current in a loop. Notice the direction of the (positive) current, it flows out of the zinc and into the steel. When steel corrodes (oxidizes or dissolves) it converts to Fe2+ ions that must leave (flow out), but here the current flow is inward, so any free Fe2+ ions are driven back and replate on the steel. The zinc dissolves, but Fe2+ ions are prevented from leaving, so the steel does not corrode. For larger structures where more current (amps) are needed a power supply is inserted in series with the short to raise the voltage, but the principle is the same.
Cathodic protection is shown to be a zinc/steel/seawater
battery that is 'shorted'
At anode: current is zn2+ ions as zinc dissolves
At cathode: current is OH- ions released as oxygen
in water is reduced to OH-
For current to flow in a loop there must be ionic flow at both metals. At the zinc it is carried by positive zn2+ ions that go into solution as the zinc dissolves. At the steel it is carried by negative OH- ions released from the steel into solution. Oxygen (dissolved gas in seawater) is relatively easily reduced to OH- ions (in basic electrolyte). Since the reaction at the steel/seawater interface involves only dissolved oxygen and water, the steel is unaffected. Here is the equation:
Reducing oxygen (plus water) to OH- (in base)
O2 + 2H2O + 4e- => 4OH-
Eo=0.40V (from WebElements)
Note talk of a sacrificial anode is somewhat misleading. In passive protection an anode dissolves to generate a battery voltage that in turn drives the current, but with active protection an external power supply is used to force current to flow into the ship's metal, so here there is no dissolving metal.
Do the potentials below show why zinc provides cathodic protection for ships? (yes) "The simplest method to apply cathodic protection is by connecting the metal to be protected to another more easily corroded metal." First used for ship protection in 1824, iron was used to protect copper ship bottoms from corroding. Zinc (along with magnesium and aluminum) are used to protect steel.
Mg(s) => Mg2+ + 2e- Eo
= +2.37V
Al(s) => Al3+ + 3e-
Eo = +1.66V
Zn(s) => Zn2+ + 2e-
Eo = +0.76V
Fe(s) => Fe2+ + 2e-
Eo = +0.44V
=> Fe3+ + 3e-
Eo = +0.04V
-----------
Cu(s) => Cu2+ + 2e-
Eo = -0.34V
Pt(s) => Pt2+ + 2e-
Eo = -1.18V (unreactive, does not ionize)
All these metals, except copper and platinum, 'want' to dissolve in ionized solutions (to oxidize). The sign change at copper indicates that copper doesn't 'want' to dissolve, that copper ions in solution 'want' to plate out (to be reduced).
Steel corrosion
Steel corrosion
in sea water is actually a subtle effect. If the steel hull of ship is
not at a uniform potential (for whatever reason), then the slight differences
in voltage along the hull will drive a weak current to flow from the more
positive voltage regions through the sea water to the more negative regions.
The area of the hull with outward flowing (positive) current will gradually
thin as it is losing Fe+++ ions and/or corrode as Fe+++ ions join up with
OH- ions to form ferrous oxide.
The equations below show the flow of electrons (through the steel) to the negative steel regions allows oxygen in the water to reduce water molecules releasing OH- ions, which is what actually carries the current in the regions of the steel not corroding.
positive voltage areas 2Fe => 2Fe++ (to sea water) + 4e- (to steel) Eo = +0.44V
negative voltage areas (reduction of water to OH- using
dissolved oxygen)
2H20
+ O2 (from water) + 4e- => 4 OH- (to sea water)
Eo = +0.40V
ions combine
2Fe + O2 + 2H2O => 2Fe (OH)2
(ferrous hydroxide, corroded iron)
Cathodic protection in practice
The figure
(below) shows that a cathodic protection cell is basically a shorted
battery. Aluminum (al+++) goes into solution releasing electrons and generating
a voltage that drops across the sea water between the electrodes. Notice
the red shorting wire in the figure (between aluminum anode and steel cathode).
As in any battery, the active aluminum (anode) is the negative terminal
and the steel (cathode) is the positive terminal, but here they are shorted
so all the potential drops across the impedance of the electrolyte. The
(positive) current flow in the electrolyte, as in any battery, is anode
to cathode, the current carried by al+++ at the aluminum interface and
OH- at the steel interface.
At the steel electrons from aluminum reaction (via wire) + oxygen (from water) + water throw OH- ions into solution (water). The OH- ions will probably link up with the positive aluminum ions forming some sort of aluminum hydroxide, but who cares. The electrons coming into the steel allow oxygen to reconfigure water into OH- (hydroxide ions) at the steel surface, but this does not affect the steel.
at aluminum 'anode'
4Al => 4Al+++ + 12 e-
Eo = +1.66V
at steel cathode
6H20 + 3O2 + 12e- => 12OH-
Eo = +0.40V
ions combine
4Al + 3O2 + 6H20 => 4Al(OH)3
(aluminum hydroxide)
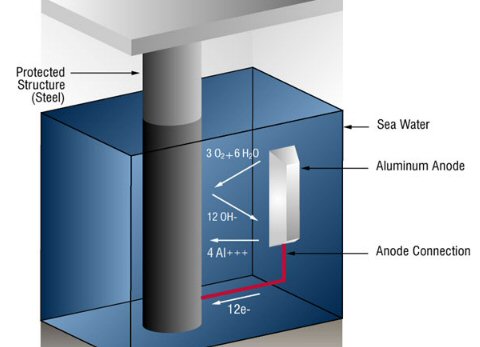
aluminum (al+++) goes into solution
electrons from aluminum reaction + oxygen (from water)
+ water => OH- ions (into water)
(left pointing arrow on [3O2 + 6H2O] just indicates
this material comes in from solution to the steel)
source --- http://www.cathodicprotection101.com/
Powered cathodic protection
Cathodic protection
can be powered by a power supply too. The principle is pretty simple. The
steel is protected if the whole surface has (positive) current flowing
into it from sea water! Positive current in is actually OH- ions
out,
where ON- ions are created at the steel interface and released into the
sea water. Clearly the power supply positive goes to the sacrificing metal
(aluminum above) and the negative to the steel. A slight twist with powered
protection is that an active dissolving metal (like aluminum) is not needed
for 'battery action', so a non-dissolving anode is usually used. In this
case the current at the anode is carried by incoming (salt) chloride ions
(cl-) from seawater being oxided to chlorine gas [2Cl- => Cl2(g) + 2e-
Eo = -1.36V], which since the oxidation potential is negative takes energy
and must be driven.
Galvanized steel
Galvanized
steel is steel coated with a thin layer of zinc for corrosian protection.
It seems funny to have a material that wants to dissolve as protection,
but obviously it works. I read that zinc protects partly by growing (from
oxidizing + CO2) a tough surface layer, and partly because nearby zinc
provides cathodic protection to scratches or exposed steel ends.
---------------------------------------------------------------------------------------------------------------------
Electrochemistry
cell
Probably the
simplest electrochemistry cell is a copper purifying cell. One electrodes
is [copper + impurities] while the other is purified copper, both in a
copper sulfate solution. Copper wants to plate out contributing 0.35V
to potential of a Daniell battery. Forcing power into the cell forces copper
to dissolve at one electrode and to plate out at the other. Since the two
electrode batteries are opposed, in principle it only takes a small external
voltage to made the reaction go.
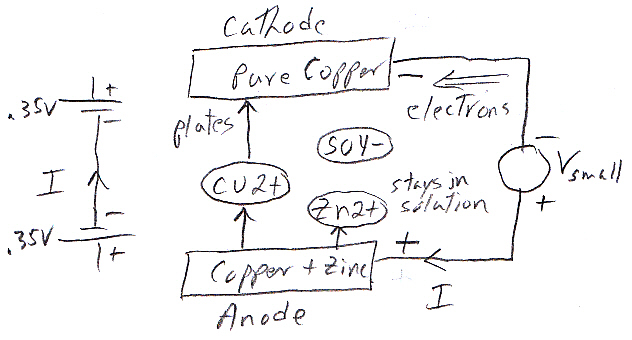
my sketch of a copper purifying electrochemistry cell
I show the external power supply voltage as Vsmall
because ideally the power (I x 0.35V) released by
copper reduction at the cathode is enough to drive
copper oxidizing at the anode.
cathode: cu2+ + 2e- => cu(s)
Eo = +0.35V
anode: cu(s) => cu2+ + 2e-
Eo = -0.35V
Notice compared to a battery 'cathode' and 'anode' terminal names are flipped. Copper ions (cu2+) go into solution at the anode and are reduced at the cathode where the electrons come in. Zinc dissolves much more easily than copper, so zinc impurities at the anode dissolve too, but it takes more energy to plate out zinc than copper, so the zinc ions stay in solution. Not sure about the sulfate ion (SO4 2-), but I read the 'sulfate ion is a very weak base, so it undergoes negligible hydrolysis in solution.' (I don't see any simple oxidation reaction for the sulfate ion in extended reduction table, most just convert it to other variants of S + O).
How do ionic conductors conduct?
I gradually
came to realize that to say a liquid conducts, as is often stated, because
it contains ions tells you nothing. Electrons don't piggy back on ions.
Seemed to me all conducting liquids had to have reducing reactions at one
electrode (to accept electrons) and oxidizing reactions at the other to
disgorge them. For example, when I thought in those terms I realized I
had no idea how a salt (sodium cloride) solution conducts. Confirming what
I suspected, I found this (excellent Keil University lecture series):
-- In contrast to purely electronic current transport, there is always a chemical reaction tied to the current flow that takes place wherever the ionic current is converted to an electronic current - i.e. at the contacts or electrodes.Current through salt (NaCl)-- We used the simple relation that d (lnc(x)) / dx = 1/c(x) · dc(x)/dx. This little trick makes clear, why we always find relations between a voltage and the logarithm of a concentration. This is a kind of basic property of ionic devices. It results from the difference of the driving forces for the two opposing currents as noted before: The diffusion current is proportional to the gradient of the concentration whereas the field current is directly proportional to the concentration.
-- The electrochemical potential thus is a real energy like the potential energy or kinetic energy.
-------------
2Cl- => Cl2(g) + 2e- (anodic reaction)
2H2O + 2e- => 2OH- + H2(g) (cathodic reaction)
2NaCl + 2H2O => Cl2 +2NaOH + H2 (overall reaction)
-------------
2Cl- => Cl2(g) + 2e-
Eo = -1.36V
2H2O + 2e- => H2(g) + 2OH-
Eo = ???
OH- + Na+ => NaOH
Eo = ??
OH- + H+ => H2O
E0 = +?? exothermic
Na+ + e- => Na(s)
Eo = -2.71V
reduction of H+ to hydrogen gas
2H+ + 2e- => H2(g)
Eo = 0.0V
oxidation of water to oxygen gas
Anode (oxidation) 2H2O
=> O2(g) + 4H+ + 4e- Eo = -1.23V
oxidation of OH- to oxygen gas
4OH-(aq) => O2(g) + 2H2O + 4e- Eo = -0.40
Combined either half reaction pair yields the same overall
decomposition of water into oxygen and hydrogen:
Overall reaction:
2 H2O => 2H2(g) + O2(g)
2Na (in Hg) + 2H2O => H2 +2NaOH + Hg
Na+ + e- => Na
Eo = -2.71V
Running current (electrolysis) through molten sodium chloride (Downs process) reduces Na+ to sodium metal and oxidizes Cl- to Cl2 (clorine gas). So at least when driven hard (conducting a lot of current and with a substantial overvoltage), positive zinc ions are reduced to zinc metal and negative cloride ions are oxidized to chlorine gas. (so what happens at low voltages and currents?)
Following materials are produced by electrolysis: Aluminium (extraction) · Calcium metal · Chlorine · Copper · Electrolysed water · Fluorine · Hydrogen · Lithium metal · Magnesium · Potassium metal · Sodium metal · Sodium hydroxide · Zinc
Looking for sodium and chlorine potentials in extended standard reduction table I find. These agree (exactly) with the Downs cell reaction used to convert molten NaCl (oxidize Cl- and reduce Na+) to sodium and chlorine gas. Wikipedia says theoretically the reaction should go at 4V [1.36V chlorine + 2.71V sodium = 4.07V], but in practice it takes 8V.
So how does a salt solution conduct? I was hoping to see some other sodium or chlorine reactions in the extended reduction tables, but nope, not there. Here's a hint (from below)
-- However, with salts of some metals (e.g. sodium) hydrogen is evolved at the cathode. (This is because reduction of H+ (0V) occurs at a much lower voltage than Na+ (-2.71V). Sodium ions are very difficult (take a lot of energy) to reduce to sodium metal.
-- For the electrolysis of neutral (pH 7) sodium chloride, the reduction of sodium ion is thermodynamically very difficult and water is reduced evolving hydrogen leaving hydroxide ions in solution. At the anode the oxidation of chlorine is observed rather than the oxidation of water since the overpotential for the oxidation of chloride to chlorine is lower than the overpotential for the oxidation of water to oxygen. The hydroxide ions and dissolved chlorine gas react further to form hypochlorous acid.
as cell operates NaOH in solution grows
Words on sodium chlorie electrolysis are: "Water is reduced at the cathode and hydrogen gas is evolved. The remaining hydroxide ion combines with the sodium to form sodium hydroxide." Sodium hydroxide (NaOH) is very soluble in water with liberation of heat. So the equations must be
2Na+Cl + 2 H2O + 2 e => H2(g) + 2 Cl + 2 NaOH (Cl- is oxidized to Cl(g) at the anode)
Sodium hydroxide (base) + hydrocloric acid yields a salt (sodium chloride) and water. This type of reaction with a strong acid releases heat, and hence is referred to as exothermic
NaOH(aq) + HCl(aq) => NaCl(aq) + H2O(l)
In general such neutralization reactions are represented
by one simple net ionic equation:
OH-(aq) + H+(aq) => H2O(l)
Ep = +??
---------------------------------
Electrolysis (Wikipedia)
-- Using a
cell containing inert platinum electrodes, electrolysis of aqueous solutions
of some salts leads to reduction of the cations (e.g., metal deposition
with, e.g., zinc salts) and oxidation of the anions (e.g. evolution of
bromine with bromides). However, with salts of some metals (e.g. sodium)
hydrogen is evolved at the cathode, and for salts containing some anions
(e.g. sulfate SO4 2-) oxygen is evolved at the anode. In both cases this
is due to water being reduced to form hydrogen or oxidised to form oxygen.
In principle the voltage required to electrolyse a salt solution can be
derived from the standard electrode potential for the reactions at the
anode and cathode. The standard electrode potential is directly related
to the Gibb's free energy, delta G, for the reactions at each electrode
and refers to an electrode with no current flowing. An extract from the
table of standard electrode potentials is shown below.
I reversed the order (as shown in Wikedia -- electrolysis) to make all the potentials negative, because this is the best way to think about electrolysis where power is being put in (generally to both electrode reactions).
Na+ + e- => Na(s)
Eo = -2.71V
Zn2+ + 2e- => Zn(s)
Eo = -0.76V
reduction of H+ to hydrogen gas
2H+ + 2e- => H2(g)
Eo = 0.0V
2Br => Br2(aq) + 2e-
Eo = -1.09V
oxidation of water to oxygen gas
2H2O => O2(g) + 4H+ + 4e- Eo = -1.23V
2Cl- => Cl2(g) + 2e-
Eo = -1.36V
2SO4 2- => S2O8 2 + 2e-
Eo = -2.07V
reduction of water to OH- (using dissolved oxygen)
2H2O + O2(g)
+ 4e- => 4OH-(aq)
Eo = +0.40
Electrolsis of water
reduction of water at cathode
"To add half reactions they must both be balanced with either acid or base."
4H2O + 4e- => 2H2(g) + 4OH-(aq)
Anode (oxidation)
2 H2O => O2(g) + 4H+(aq) + 4e-
Combined either half reaction pair yields the same overall
decomposition of water into oxygen and hydrogen:
Intermediate:
4OH- + 4H+ => 4H2O
Overall reaction:
2 H2O => 2H2(g) + O2(g)
-- It is more difficult to reduce sodium ion to sodium metal than it is to reduce zinc ion to zinc metal, and it is more difficult to oxidise sulfate anions than it is to oxidise bromide anions.
electrolysis of water (cont)
-- Care must
be taken in choosing an electrolyte, since an anion from the electrolyte
is in competition with the hydroxide ions to give up an electron. An electrolyte
anion with less standard electrode potential than hydroxide will be oxidized
instead of the hydroxide, and no oxygen gas will be produced. A cation
with a greater standard electrode potential than a hydrogen ion will be
reduced in its stead, and no hydrogen gas will be produced.
-- The following cations have lower electrode potential than H+ and are therefore suitable for use as electrolyte cations: Li+, Rb+, K+, Cs+, Ba2+, Sr2+, Ca2+, Na+, and Mg2+. Sodium and lithium are frequently used, as they form inexpensive, soluble salts.
-- If an acid is used as the electrolyte, the cation is H+, and there is no competitor for the H+ created by disassociating water. The most commonly used anion is sulfate (SO4 2-), as it is very difficult to oxidize, with the standard potential for oxidation of this ion to the peroxodisulfate ion being -2.05 volts.
-- Strong acids
such as sulfuric acid (H2SO4), and strong bases such as potassium hydroxide
(KOH), and sodium hydroxide (NaOH) are frequently used as electrolytes.
====================================================
Understanding H2O <=> O2 (Wiki 'redox')
-- Cellular
respiration is the oxidation of glucose (C6H12O6) to CO2 and the reduction
of oxygen to water.
C6H12O6 + 6 O2 => 6 CO2 + 6 H2O
-- Photosynthesis
involves the reduction of carbon dioxide into sugars and the oxidation
of water into molecular oxygen.
6 CO2 + 6 H2O + light energy => C6H12O6 + 6 O2
in acid?
O2 +
4e- + 4H+ => 2H2O reduction of oxygen
Eo=+1.23V spontaneous
2H2O => 4H+
+ O2 + 4e- oxidation of water
Eo=-1.23V electrolysis
2H2O
+ 2e- => H2 + 2OH-
Eo=-0.83V
To be oxidized is to lose an electron or hydrogen atom
To be reduced is to gain an electron or hydrogen atom
Eo (@ standard conditions) is the electrochemical version
of free energy
Importnat biological production of methane is by acetate methanogensis.
CH3C00- + 2H2O => 2CO2 + 8H+ + 8e- (not balanced inH!)
CO2 + 8e- + 8H+ => CH4 + 2H2O
---------------------------------------------------
CH3C00- => CO2 + CH4
thermodynamicly favored but very
slow without microbial assistance
Methanogenesis
4H2(g) + CO2 => CH4 + 2H2O
which I separate as
4H2(g) => 8H+ + 8e-
CO2 + 8H+ 8e- => CH4 + 2H2O
-------------------------------------------------------------------------------------------------------------
Mysteries
High
electrolyte potential mystery
In learning
a new topic I often come across a mystery, a conflict that is hard to resolve,
and here the mystery was the claim that the electrolyte potential was high,
really high, higher than both terminals.
Early on I came across an introductory college lecture on batteries that showed metals in contact with electrolytes tend to develop large (Nernst type) space charge layer with an E field pointing toward the metal to limit diffusion of the metal into the electrolyte. So far, so good, but the lecture (figure below) showed these space charge layers push the potential of the electrolyte way up, several volts more positive than either terminal! This was seemingly confirmed by Wikipedia, which said electrolyte potential was not easily measured, but that the reference hydrogen electrode voltage was +4.44V, and when I later leaned that Standard Reduction voltages range from -3V to +3V, a +4.44V offset made them all positive, so it did look like the electrolyte in batteries was (or could be) indeed highly positive.
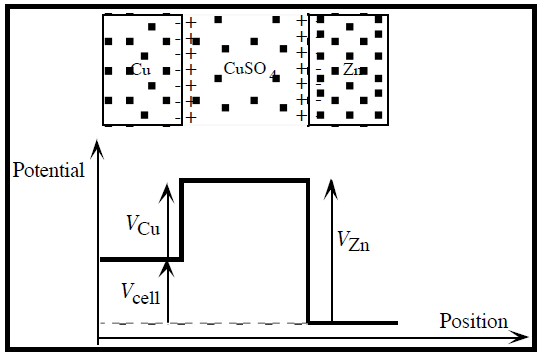
highly positive electrolyte
(Univ of Syndey battery lecture)
But from a circuit model or energy viewpoint a highly positive electrolyte in a battery makes no sense. Ionic currents going uphill and downhill through high space charge potentials would mean large power flows are occurring continuously within the battery (between electrodes), and there was not a hint from battery application literature that anything like this was happening.
I drew my battery circuit models using Standard Reduction voltages (ignoring the high potential electrolyte arguments) and everything worked. The circuit models showed the potential of the electrolyte was between terminal voltages. For weeks I was bothered by this electrolyte potential conflict, but was unable to resolve it.
Resolution of mystery
I now think the
high electrolyte potential arguments are either just plain wrong or are
an irrelevancy. It might be that in some batteries at zero current
(no load) that the potential of the electrolyte tends to float up a little.
It's like having two small capacitors in series and asking what is the
voltage in the center. The key is that even if the potential across the
electrode space charge layer capacitances floats up (due to a small charge
flow) they are going to be discharged (essential the capacitance gets shorted)
when any significant current starts to flow from the battery. The key reason
is this:
A voltage higher than the chemical reaction generates, its Standard Reduction/Oxidation potential, cannot be sustained across a space charge layer with current flowing across it, because there is no source to generate the 'extra' power [P = I x (Vreaction + Velectrolyte float)].Hence, it's totally unimportant what the voltage of the electrolyte is in a battery with open terminals. What's important is that as soon as the battery begins to work the electrolyte potential is well determined by local electrode batteries based on the Standard Reduction potentials for the reactions. It is deeply misleading to beginning students, as this college lecture did, to dwell on the (possible) high potential of the electrolyte (at no current), as though this was an important concept to understand, when it fact it is an irrelevant detail.
Zinc sulfur mystery
I though I
knew how the Volta zinc/copper/sulfuric acid cell worked. But then I see
on YouTube that zinc dumped into sulfuric acid in a few seconds releases
a lot of heat boiling the liquid with a huge amount of hydrogen bubbling
out. The current from the dissolving zinc+ ions is cancelled by H+ ions
in the acid moving to the zinc and being reduced.
So the mystery is this: Why doesn't this happen in zinc/copper/sulfuric acid cell? The zinc is in contact with the sulfuric acid. I don't see how adding in some copper to the solution, open circuit, can make any difference. Is it that the reaction rate depends on the concentration of sulfuric acid. But there's a huge range to span from seconds to days/week/month?? for a volta pile. It it possibly that the reaction rate is limited physically (by diffusion). The voltage pile uses cardboard soaked in sulfuric acid. But in the tutorial figure above zinc and copper strips are just sitting in sulfuric acid. Is there a thershold effect where maybe zinc moves into dilute sulfuric acid, the local H+ react, but then the zn+ screens the negative charge on the zinc, so no further H+ diffuse in?
Zinc with (pure) sulfur is really reactive. It makes flash explosions (6 parts zinc to 1 part sulfur) and even rocket fuel! There are a lot of zinc/sulfer rockets on YouTube.
There's a YouTube
video showing this highly exothermic reaction, the acid getting hot and
hydrogen gas bubbling off vigerously --- http://www.youtube.com/watch?v=3ijmhYXpBBM
10% sulfuric
acid, 90% water (still pretty vigerous)
http://www.youtube.com/watch?v=ZrifMySH6zI&feature=related
-- Penny is
zinc with copper coating. Scratch a penny and dump in 10M HCL (muriatic
acid). In 45 minutes all the zinc has dissolved into the acid with H2 &
maybe HCl released and what's left is a copper shell. Hcl does not react
with copper.
http://www.youtube.com/watch?v=6ZM5IOgWmJs&feature=related
Why
are zinc and copper so different?
Zinc (if you
bleed away it's electrons) 'wants' to change from solid to ions in solution,
while copper (if you provide it with electrons) 'wants' to do the reverse,
to change from ions in solution to solid copper. This difference makes
copper a good mate with zinc in batteries, because its local battery polarity
is reversed from zinc, so the Eo of the two reactions add.
The mystery (of which I have seen zero explanation) is why are these two metals so different in their standard reduction potentials? Zinc and copper are adjacent on the periodic table (#30 and #29) and their electronegativities are pretty close too (1.65 and 1.90). What atomic difference explains this? Here's some data (from Wikipedia) for zinc and copper along with others in their periodic table groups.
gold
#79 ...5d106s1 9.22
eV (1st ion pot) 0.130 eV heat of fusion
1,064 C melt 22.1 nohm-m
silver #47
Kr 4d105s1 7.58 eV (1st ion pot) 0.117
eV heat of fusion 962 C melt
15.9 nohm-m
copper #29
Ar 3d104s1 7.73 eV (1st ion pot) 0.137
eV heat of fusion 1,085 C melt 16.5 nohm-m
zinc
#30 Ar 3d104s2 9.39 eV (1st ion pot)
0.076 eV heat of fusion 420 C melt
59 nohm-m
cadmium #48 Kr 4d105s2
8.99 eV (1st ion pot) 0.064 eV heat of fusion
321 C melt 73 nohm-m
mercury #80
...5d106s2 10.4 eV (1st ion pot) 0.024 eV
heat of fusion -39 C melt
961 nohm-m
 .
. 
Size of ball indicates: melt temperature (left), ethalpy
(energy) of fusion (right)
Zinc, cadmium, mercury are on the right (of metal,
copper color)
2nd from right is copper, silver, gold
(source -- http://www.webelements.com/periodicity/melting_point)
What jumps out from the numbers is that zinc family has much lower melting temperatures and much lower heats of fusion than the copper family. The two periodic ball charts above, where the size of the ball indicates the value, are for melt temp and (energy) of fusion. Zinc is in the upper right hand corner of the normal metals, which are shown copper colored on the charts. Note the melt and heat of fusion ball charts show pretty much the same pattern for metals. These charts show the zinc family (zinc, cadmium, mercury) have significantly lower melt temperatures and heat of fusion than all the other 28 (normal) metals.
Melting temperature, heat of fusion, lattice energy, (standard reduction potential?) are all related (I think) to the strength of atomic bonding between the metal atoms. Also I read that in principle heat of fusion is related to solubility.
Good elment reference with some nice comparison charts.
http://www.webelements.com/zinc/thermochemistry.html
Finally a clue --- zinc atoms are loosely bound with
d10s2 outer shells like mercury
I finally
found a clue about why zinc and copper are so different. In general metals
atoms are tightly bonded to each other leading to high melting temperatures
(copper is 1,085 C and tungsten is very high at 3,422 C), but three metals
are a "remarkable exception" (says Wiki 'metallic bonding'): zinc, cadium
and mercury. They are all in the same group with a full 'd10' shell under
a 's2' outer shell (a pair in the spherical 's' oribital with opposite
spins).
The result is that they have much weaker forces between their metal atoms. They are more volatile and have lower melting temperatures. They are a "remarkable exception " exception to the high temperature and tight atomic bonding of other metals (says Wikipedia, 'metallic bonding'). Zinc is 4s2 and melts at 420 C, cadmium is 5s2 and melts at 321 C, mercury is 6s2 and melts at -39 C (Only metal that's a liquid at room temperature).
-- The atoms in metals have a strong attractive force between them. Much energy is required to overcome it. Therefore, metals often have high boiling points, with tungsten (3,422 C) being extremely high.
-- A remarkable exception (to high temp melting) are the elements of the zinc group: Zn, Cd,and Hg. Their electron configuration ends in ...s2 and this comes to resemble a noble gas configuration like that of helium more and more when going down in the periodic table because the energy distance to the empty 'p' orbitals becomes larger. These metals are therefore relatively volatile. Zinc for example is unsuitable for use in ultra high vacuum applications.
-- With the advent of electrochemistry it became clear that metals generally go into solution as positively charged ions and the oxidation reactions of the metals became well understood in the electrochemical series. A picture emerged of metals as positive ions held together by an ocean of negative electrons.
-- Metals are insoluble in water or organic solvents unless they undergo a reaction with them. Typically this is an oxidation reaction that robs the metal atoms of their itinerant electrons, destroying the metallic bonding.
Where is the Anode?
It's a detail
and not really that important, but initially I was surprised to find that
(nearly) every reference was labeling the negative battery terminal the
'anode'. As a working EE I had always thought (or perhaps just assumed)
that the positive terminal of a battery was the anode. In my defense Wikipedia
(Anode) does point out that
a) There is
a widespread misconception is that anode polarity is always positive (+),
and
b) Some USA
battery companies call the positive terminal the anode, and that this definition
has the virtue that the anode doesn't jump around.
Real reasonThis is a curious definition. It means for a battery discharging that the anode is the negative terminal, but for a battery charging the anode is the positive terminal! What, the anode moves? Yup, the consensus definition of 'anode' means the anode and cathode of a rechargeable battery flips when it shifts from charging to discharging. It means that the 'anode' of your car battery is continually jumping back and forth between its terminals, because the anode definition is keyed to the direction of current flow!
It finally came to me though the real reason why I always thought the positive terminal of batteries was the anode. In diodes and LED's, devices I designed with for years, the positive terminal is called the anode. In fact the positive terminal of diodes on datasheets is often marked with an 'A'. In the general case Wikipedia notes (article, Anode): The anode is the terminal of a polarized electrical device into which current flows. This applies to diodes and batteries (both discharging and charging).From another point of view when power is flowing into a device the positive voltage terminal is the anode. For example, diodes and charging batteries. When power is flowing out of a device, like a discharging battery, the negative terminal is the anode.
The anode is where the oxidation reaction takes place, where the electrons come out, where the (positive) current flows in. I have adapted to it, and from an internal electrochemistry point of view it's clean, but for a rechargeable battery it is a little weird.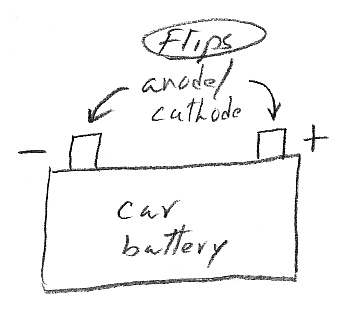
Anode/Cathode 'labels' of a car battery terminals flip back and forth
depending on whether the battery is charging or discharging.
This is a slightly weird byproduct of defining the 'anode' as the terminal where the current comes in.
Discharge: anode/cathode
Charge: cathode/anode
For fun I went looking for an example of the positive terminal marked 'anode', and thought I had found it in a silver oxide cell (below, left) in a Radio Shack online battery guide. I just assumed that the small top terminal of a button battery was positive, like it is in a flashlight C or D cell. But this is wrong! In fact the small terminal of many button batteries, like silver oxide and manganese dioxide cells, is the negative terminal, and the case is the positive terminal. When I looked at a manganese oxide button battery, I found a big '+' on the bottom of the case.
 .
. 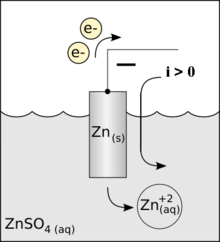
Radio Shack sketch of silver oxide (primary) cell
Wiki (Anode) -- 'Anode' battery terminal
(downloaded 9/10, copyright 1990)
is negative metal (zinc) can
http://support.radioshack.com/support_tutorials/batteries/batgd-B01.htm
Anode/cathode confusion
My first battery
sketches labeled the + terminal (non-metal) 'anode'. Wikipedia and
other references show I have the names anode/cathode reversed, they
say the cathode is the + (non-metal) terminal. Then I find this in Wikipedia
-- Anode
-- In
the United States, many battery manufacturers regard the positive electrode
as the anode, particularly in their technical literature. Though technically
incorrect, it does resolve the problem of which electrode is the anode
in a secondary (or rechargeable) cell. Using the traditional definition,
the anode switches ends between charge and discharge cycles.
Yikes! In
other words there is no agreement on which terminal should be labeled 'anode'
and which 'cathode'! The standard anode/cathode definition is ridiculous,
see below. It's not physical it's a function of current flow, so it 'flips'
when the cell is charged. (slightly) ridulous.
-- In a recharging
battery, or an electrolytic cell, the anode is the positive terminal, which
receives current from an external generator. The current through a recharging
battery is opposite to the direction of current during discharge; In other
words, the electrode which was the cathode during battery discharge becomes
the anode while the battery is recharging. (wiki anode)
I checked
Energizer (dry cell) does call the zinc terminal the 'anode'
Everyready has the metal (zinc) as the anode
-- Anodes are the electrodes that are oxidized (give up electrons)
Silver oxide button battery case is marked '+'
One book refererence
calls positive ionic current flow (from anode) 'anodic" and negative ion
current flow the cathode 'cathodic' and adds that the terms anodic and
cathodic are strictly related to the direction of current flow. (update)
Have never again seen this terminology.
=========================================================
Metals
and non-metals in periodic table
I found the
type of periodic table that Prof Sadoway had on screen with color coded
metals and non-metals.

purple (here 'semi-conductors') are more generally
called 'mettaloids'
(screen captured from http://www.lenntech.com/periodic/periodic-chart.htm)
Metals
It is a remarkable
fact that the overwhelming majority of elements in the periodic chart are
metals. Above metals are color coded dark blue, but the two bottom row
(lanthanides and actinides) are metals too. All these elements want to
donate electrons. In pure form they look metalic (or silvery), lustrous,
and many are soft and ductile. Their metalic appearance is due to the fact
that light is reflected from the many free electrons near the surface.
All these loosely bound electrons means metals have high electrical conductivity,
positive valence, and in solution they readily lose electrons forming positive
ions (cations).
Non-metals
All the non-metal
elements are in the right hand corner of the periodic chart (with the exception
of hydrogen): carbon, nitrogen, oxygen; phosphorus, sulfur; plus hydrogen
and the halides (florine, chlorine, bromine, and iodine), which are missing
one electron from their outer shells. Non-metals are electron absorbers
with high electronegativity and negative valence (they want electrons).
In solution they readily accept electron(s) forming negative ions. In batteries
the metal goes at the one electrode (negative) and a non-metal at the other
electrode (positive).
Metalloids
In between the metals
and non-metals are a few elements (color coded purple) that can either
accept or donate electrons, known as semiconductors or (more recently)
metalloids. Two of the metalloids, antimony (#51) and tellurium (#52),
which are both silvery and lustrous looking like metals, are used in the
liquid battery.
Two 'metal' batteries --- copper & zinc---------------------------------------------------------------------------------------------------------------------
When you begin to look at real batteries, you see that the metal/non-metal view is a simplified viewpoint. A classic, very early, battery is just a strip of zinc and copper inserted into an electrolyte, and a two electrolyte version (zinc in zinc sulfate and copper in copper sulfate) is the Daniell cell from 1836, which is often used in classroom battery demos.Where is the non-metal? Clearly in the atomic, metallurgical sense both zinc and copper are metals. In fact copper (#29) zinc (#30) are both adjacent in the periodic table and are toward the right side being elements 11 and 12 in a row of 18. Since zinc is more to the right, a guess would be it acts as the non-metal, but this is wrong. The electronegativities are reversed from the normal pattern with (left side) copper having the higher value (1.90) and (right side) zinc the lower (1.65). Hence it is zinc that acts as the 'metal' releasing electrons and is the negative terminal of the battery.
Standard concentration
Voltages from
nernst equation are usually figured as two factors: reference value plus
a correction for concentration. The concentration adjustment voltage is
figured for each factor of 10 in concentration ratio (at room temperature)
from the 'standard concentration' as 58 mv [= 25 mv x ln (10)]. But how
concentrated is the standard concentration? I see 'standard concentration'
(1.00M) variously listed as [1 mole/liter] or [1 mole/kg], which for water
are the same.
mole of water
18 grams (O is atomic weight 16 + 2
hydrogen)
weight of 1 liter of water
1,000 gram (1 kg/L)
concentration (1,000 gm/18 gm)
55 mol/liter
Hence a 'standard concentration' (for water in water!) appears to be 1.8% (=18gm/1,000gm). A more practical case is salt dissolved in water. Molecular weight of NaCl is 58, so a 1M concentration of salt (in water) is 58 grams of salt dissolved in 1 liter (total), which has a weight of water (I think) still about 1,000 grams, so here the 1M salt solution is 5.8% solute.
Sulfuric acid (H2SO4) has a molecular weight of (32 + 4 x16 +2 = 98) or x5.4 heavier than a water molecule (18). If the volume occupied by a molecule of (liquid) H2SO4 was the same as water, a liter of sulfuric acid would weight x5.4 more than water (or 5.4 kg/L), but in fact the density of pure sulfuric acid is (about) 1.8 kg/L. So the volume occupied by a molecule of sulfuric acid (H2SO4) must be about x3 (= 5.4/1.8) the volume of a water molecule (H2O). (This seems reasonable since the 3rd root of 3 is 1.44.) With three times less molecules than water per liter, the mol/L of sulfuric acid is 18 (= 55 mol/L water x 1/3rd molecules).
PH
Ph is a logrithmic
measure of the concentration (meaning the ratio of solute
to solvent) of H+ ions in solution. (pneumonic for 'ph' is 'power of hydrogen')
ph = -log (H+)
or for acids in water (where H+ attaches to H2O forming H3O+)
ph = -log [H3O+]
More accurately ph is (approximately) the negative of the logarithm (base 10) of H3O+ (hydronium ions). Water has a ph=7 and is taken as neutral. So water with ph 7 has a molar concentration of 0.0000001 or 10^-7, meaning for every 10 million water molecules there is one (effective H+ion, which is really H+ glommed onto H2O to make H3O+ ion). A typical acid is H2SO4, which in water ionizes to H+ and HSO4- ions.
A solution of a strong acid can have a pH near 0 (1 mol/L) or somewhat negative. Acid with a concentrations of 10 mol/L has a negative ph (ph = -1) (Above I estimate that sulfuric acid pure will be about 18 mol/L).
Faraday's two laws of electrochemistry
1) The amount
of a substance deposited on each electrode of an electrolytic cell is directly
proportional to the amount of electricity passing through the cell.
2) The quantities
of different elements deposited by a given amount of electricity are in
the ratio of their chemical equivalent weights.
Walther Nernst
In 1889, Nernst
elucidated the theory of galvanic cells by assuming an "electrolytic pressure
of dissolution," which forces ions from electrodes into solution and which
was opposed to the osmotic pressure of the dissolved ions.
Standard reduction potential
The more
positive
the reduction potential the more easily a species is reduced (the more
reactive it is an an oxidizing agent). If it is negative, it cannot be
reduced by hydrogen (zero voltage reference reaction where hydrogen gas
is oxidized (gives up its electron) to the +H ion).
In fact the 'Standard reduction potential' is measured by setting up a 'battery' (two half reactions connected by a salt bridge) with hydrogen gas acting as the metal (releasing electrons as H+ go into solution) and the material under test as the non-metal, accepting the electrons and being reduced. (Hydrogen electronegativity = 2.20.) Oxidizing portentials (losing electrons) are just the negative of these reduction potentials.
Standard reduction potential
electronegativity
------------------------------------
---------------------
lithium
Li+(aq) + e- => Li(s)
-3.05 V
0.98
potassium
K+(aq) + e- => K(s)
-2.93 V
0.82
zinc
Zn2+(aq) + 2 e- => Zn(s)
-0.76 V
1.65
hydrogen
2H+ + 2 e
=> H2(g)
0.0 V
2.2
copper
Cu2+(aq) + 2 e- => Cu(s)
+0.34 V
1.90
silver
Ag+(aq) + e- => Ag(s)
+0.80 V
1.93
fluorine
F2(g) + 2e- => 2F-
+2.87 V
3.98
Long table of standard reduction potentials (in alphabetical
order)
http://web.archive.org/web/20070518092613/http://www.northland.cc.mn.us/Chemistry/standard_reduction_potentials.htm
Electronegativity
First thing that
strikes me here is that electronegativity is only loosely correlated to
standard reduction potential.
Hydrogen almost exactly in center of measured potential
range
Second is that (while
I have seen nobody say this) it looks obvious that a key reason hydrogen
has been chosen as the 0V reference point is that it is almost exactly
in the middle of the measured six voltage making it (-3V to +3V).
Sign indicates direction of power flow
Third, reduction
at an the electrode interface requires an electron flow in,
which is (positive) current flow out. Hence a positive voltage
(positive reduction potential) means power flowing out, the electrode
reaction is spontaneous and putting out power. In contrast a negative
voltage (negative reduction potential) means power flow in, the
reaction is being forced.
Measuring reduction potentialMaterials wanting to be reduced have large 'positive' reduction potentials
The relation of power flow to reduction voltage (potential) is clear if we consider the material being tested is used as the non-metal (reduction) half-cell with the hydrogen reference electrode used as the oxidizing (metal) electrode. Since the hydrogen reference electrode half-cell voltage is zero, the measured voltage, relative the hydrogen reference electrode, is the reduction potential of the material under test.
-- Thus Zn2+ whose standard reduction potential is -0.76 V can be oxidized by any other electrode whose standard reduction potential is greater than -0.76 V (eg. H+(0 V), Cu2+(0.16 V), F2(2.87 V)) and can be reduced by any electrode with standard reduction potential less than -0.76 V (eg. H2(-2.23 V), Na+(-2.71 V), Li+(-3.05 V)). (Wiki - standard electrode potential)
Translation ---- Zinc will be the negative terminal (metal releasing electrons) in a cell with any materials below it in the table, those with a standard reduction more positive (like copper). If zinc is used in a cell with materials above it (like lithium at -3.01V), the lithium acts as the metal (negative terminal) donating electrons and zinc would be the positive terminal accepting electrons.Cell voltage is the difference in the standard reduction potentials. For example, the zinc/copper battery has a 1.10V EMF. This can be modeled as two local batteries in series one across each terminal/electrolyte interface. The local battery at zinc terminal puts zinc -0.76V (relative to electrolyte), and the local battery at copper terminal puts copper +0.34V (relative to electrolyte). The two batteries in series add to 1.1V with zinc the negative terminal and copper the positive terminal. Common watch silver oxide watch batteries are really silver zinc batteries and have a nominal voltage of 1.5V [from above 1.56V = 0.80V - (- 0.76V)].
I read the voltage of the electrolyte (in a battery) is not really measurable, but it seems to me the above 'Standard reduction potential' can be thought of as the voltage of that terminal relative to the electrolyte. This certainly works for the zinc/carbon cell.
In the Sydney lecture the source of the 'Standard reduction potential' is given as an (effective) nernst voltage generated by the ratio of the (very high concentration) of atoms in a solid relative to the low concentration of the its ions in solutions as it dissolves. In other words it's a voltage self generated by +metal ions (charge) moving into solution to build up an E field to prevent further (net) diffusion ('dissolving') of the solid into the liquid. Sketches in the Sydney lecture (see below) show at the interface a net +metal ion layer in solution and a net - charge at the edge of the solid metal electrode. The electrode voltage is clearly driven negative relative to the electrolyte and this E field is the source of the local interface battery.
Is this right?Absolute electrode potential
But if this 'standard potential voltage' is entirely a nernst diffusion stopping voltage, I don't see how voltages of 1 to 3 volts can be generated. The voltage is supposed be relative to a standard concentration, but this looks to be 2% or so, so how can such high voltage be generated if it takes a x10 concentration factor for each 58mv!).
The clear implication here is that the electrode is positive relative to the electrolyte, but in fact the Sydney lecture shows the opposite. It shows at both the anode and cathode +ions dissolving and forming a + space charge layer in the electrolyte next to the interface with a negative layer opposite in the electrode metal. This makes an E field at both electrodes pointing from the solution to the electode, in other words both electrodes are negative relative to the electrolyte.
Spontaneous
E°cell
= E°cathode - E°anode
where E is the value in the standard reduction potential table
If E°cell > 0, then the process is spontaneous (galvanic cell)
If E°cell < 0, then the process is nonspontaneous (electrolytic
cell)
In a zinc/copper cell zinc is the metal anode (electron releasing negative terminal) with copper the positive cathode terminal. Hence [E°cell = E°cathode - E°anode] results in Ecell = +.34V (copper cathode) - (-0.76V) (zinc anode) = +1.1 V. It's positive so it's spontaneous, it can supply energy, it's a battery.
However, if I understand correctly though a material can be either anode or cathode depending on what it's paired with. So (it seems to me) we get the interesting result than any two materials in the table can be paired and we get a battery! The electrons want to move from the electron releasing material (metal) the accepting material (effective) non-metal, and in doing so energy becomes available externally. (Zinc/copper is a battery, but so (in principle) is lithium/zinc.)
Gibbs free energy
There are
actually two terms that contribute to whether or not a reaction will be
spontaneous. A reaction will go if the change in 'gibbs free energy'
has the right sign (don't know now and won't worry about what the sign),
but gibbs free energy is the sum of two terms. (Reactions go at high temp,
so I can see from below that G negative is what makes a reaction
go.)
[gibbs
free energy (kj/mol)] = [enthalpy (kj/mol)] - [temp kelvin (K) x entropy
(kj/mol-K)]
G = H - TS
H is enthalpy --a measure of the total energy of a thermodynamic system
S is entropy -- a measure of disorder (# of possible states)
G is gibbs free energy -- negative change is an exothermic reaction
positive change is an endothermic reaction
T is degrees kelvin (298 K is 25C room temperature)
Entropy goes as disorder or the number of possible states of a system. I suspect in many (most?) chemical reactions there is no change in entropy so changes in enthalpy controls. What's relevant to batteries is that when a solid dissolves into an electrolyte, entropy (S) increases. (I saw this in a chemistry textbook). An increase in S, scaled by -T, tends to drive G negative, thus tending to make the reaction go. (At this point I have no idea what the scaling is, I am going to have to do some reading up on entroy increases when solids dissolve.)
Entropy
Thermodynamically
change in S is change it heat flow (delta Q) divided by temperature. Classic
example is change in entropy of ice (273K) in a glass melting sitting at
room temperature (298K). Delta Q flows from 298K room to melt ice at 0C
(273K). Entropy of ice in glass goes up by [delta Q/273K], and entropy
of room goes down by [delta Q/298K], so entropy of the closed
system of a room with a glass in it has gone up.
Another classic is the production of syngas, where carbon (coke) reacts with steam to form a mixture of carbon monoxide and hydrogen [C + H2O => CO + H2]. There is an entrogy increase since solid carbon ends up as a gas, but the reaction only goes when T is high enough (carbon red hot and steam) to make the 'TS' term large enough to drive gibbs free energy negative.
Solvation
Dissolving
--- Ions of a solute being surrounded by molecules of a solvent is called
'solvation' (Wikipedia has an article with the title solvation).
-- 'Enthalpy of solvation' can help explain why solvation occurs with some ionic lattices but not with others. The difference in energy between that which is necessary to release an ion from its lattice and the energy given off when it combines with a solvent molecule is called the 'enthalpy change of solution'.
-- A negative value for the enthalpy change of solution corresponds to an ion that is likely to dissolve, whereas a high positive value means that solvation will not occur.
-- It is possible that an ion will dissolve even if it has a positive enthalpy value. The extra energy required comes from the increase in entropy that results when the ion dissolves. The introduction of entropy makes it harder to determine by calculation alone whether a substance will dissolve or not. (Is the implication that entropy changes are not calculable and must be measured?)
-- For solvation to occur, energy is required to release individual ions from the crystal lattices in which they are present. This is necessary to break the attractions the ions have with each other and is equal to the solid's lattice free energy (the energy released at the formation of the lattice as the ions bonded with each other). The energy for this comes from the energy released when ions of the lattice associate with molecules of the solvent. Energy released in this form is called the 'free energy of solvation'.
-- Polar solvents are those with a molecular structure that contains dipoles. Such compounds are often found to have a high dielectric constant. The polar molecules of these solvents can solvate ions because they can orient the appropriate partially-charged portion of the molecule towards the ion in response to electrostatic attraction.
-- Standard Enthalpy of solution, defined as the enthalpy change observed in a constituent of a thermodynamic system, when one mole of an solute is dissolved completely in an excess of solvent.
-- Just as the energy of forming a chemical bond is the difference between electron affinity and ionization energy, the heat of solution of a substance is defined as the sum of the energy absorbed, or endothermic energy, expressed in positive values and unit kJ/mol, and energy released, or exothermic energy (negative value).
-- Dissolution
can be viewed as occurring in three steps:
1) Breaking solute-solute attractions (endothermic), see for instance lattice
energy in salts.
2) Breaking solvent-solvent attractions (endothermic), for instance that
of hydrogen bonding.
3) Forming solvent-solute attractions (exothermic), in solvation.
The value of the overall enthalpy change is the sum of the individual enthalpy changes of each of these steps. For example, dissolving ammonium nitrate in water decreases the temperature of the solution. Solvation does not compensate energy spent in breaking down the crystal lattice, while adding potassium hydroxide will increase it.
-- 'Enthalpy change of solution' for some selected compounds (Change in enthalpy delta H in kJ/mol in water at 25°C)
Dissolving decreases temp of solution
ammonium nitrate +25.69
potassium chlorate +41.38
sodium chloride +3.88
Dissolving Increases temp of solution
potassium hydroxide -57.61
hydrochloric acid -74.84
ammonia -30.50
acetic acid -1.51
sodium hydroxide -44.51
-----------------------
Battery chemistry basics
* Metals have
a 'sea of electrons', so the atoms of metal are (in some sense) positive
ions. Solubility of neutral metal atoms is negligibly small. In other words
metals go into solution only as (positive) ions.
* Metal dipped into a solution of its salt is a special case. Salt in solution is disassociated into positive metal ions and (salt) negative ion (Cu2+ + SO4 2-). Electrical conductivity of metals shows that they consists (at least partially) of positive metal ions + electrons (Cu2+ + 2e-). In this case there is one (just one) common element in the two phases (solution and metal), so a metal ion (Cu2+) equilibrium must develop. This differs from the ordinary case of solubility because here the only equilibrium possible metal ions between two phases. Nernst thinks of this equilibrium as a balance between 'solution pressure' and 'osmotic pressure (to deposit out)'. This explains (sort of) as to why electrons don't go into solution.
* Only a minute number of ions need go into (or out of) solution to set up the electric field that exist right across the interface and which supports the potential difference between the metal and solution. The potential is defined as the work (eV) to take a unit positive charge from interior of solution to the interior of the metal.
* Nernst models
the work of dissolving as a three stage expansion process: metal ions withdraw
from the metal at electrolytic solution pressure of the metal 'P', expand
isothermally to the osmotic pressure 'p', and condense at this pressure
into solution, which he calculates as [RT ln ('P'/'p')] per mol. At equilibrium
the net work is zero, so this must match the work of a mol of charge crossing
the E field [nF x V], leading to the nernst equation:
V = (RT/nF) ln ('P'/'p')
For the voltage difference between two electrodes in the same solution 'p' drops out so the equation becomes:
delta V = (RT/nF) ln (P1/P2)
and at 25C and conversion from 'ln' to 'log' the equation becomes
delta V = (59 mv/n) log (P1/P2)
Data from tests of sulfuric acid at various concentrations show that ideal of [delta 59 mv per factor of ten] is only an approximation. The results are [20 mv to 23 mv per factor of ten] over a hundred to one range.
How does current move through an electrolyte?
A critical
thing that introductory text invariable skip over is this: How does current
flow through an electrolyte anyway? Introductory texts invariably imply
a solution will conduct if it contains mobile carriers (ions). Well, it's
not that simple. In an excellent German series of material science lectures
I found this: "In contrast to purely electronic current transport, there
is always a chemical reaction tied to the current flow that takes
place wherever the ionic current is converted to an electronic current
- i.e. at the contacts or electrodes."
Electrons don't
just jump on (and off) ions. If they do, it's a chemical reaction that
invariable emits or absorbs energy. The closest to a 'hop' is probably
a simple reaction like [zn(s) => zn2+ + 2e-] where zinc 'dissolves' into
solution as ions (zn2+) leaving behind electrons on the electrode. But
this is an exothermic chemical reaction with a standard voltage of 0.76V,
and it fact this reaction is the is the source of the 0.76V terminal voltage
in the first battery invented by Volta in 1800.
----------------------------------------
Sulfuric
acid as an electrolyte
Zinc has a
negative reduction potential (Eo = -0.76V) so it is favorable for zinc
to be oxidized (the opposite of being reduced) under standard conditions:
Zn --> Zn2+ + 2 e- Eo = +0.76V
In sulfuric acid you primarily have three species present
(not including the water):
H+, HSO4-,
and SO42-.
Of these three, H+ has the highest reduction potential:
2 H+ + 2 e- --> H2 Eo = 0.00V
So in the presence of H+ ions from sulfuric acid, zinc
will oxide to form Zn2+:
2 H+ + Zn --> H2 + Zn2+ Eo = 0.00V + 0.76V = 0.76 V
another posters summarizes: Zn(s) + H2SO4(aq) ---> ZnSO4(aq) + H2(g)
Sulfuric acid
** (summary
2) H2SO4 is a very polar molecule with strong dipole forces between molecules
that allows molecules to occasionally steal an H+ from a neighbor. In equilibrium
about 1% have an extra H+ forming positive ion H3SO4+ (effectively H+)
and 1% are missing H+ forming negative sulfate ion HSO4-, meaning about
2% of sulfuric acid molecules are ions and can conduct electricity.
This answers one question I had. Why do molecules 'come apart' into ions. Looks like the answer is that high polar molecules, like sulfuric acid, jostling about can pull off a dangling H+ for themselves. The number ionized is described by an 'autoprotolysis' equilibrium constant that this the product of the two ions.(summary) H2SO4 has a high dielectric constant (100 vs 80 for water which is also very polar, 1 for vacuum) indicating it is a very polar molecule. For reasons not really explained about 1% of sulfuric acid molecules protonate themselves deprotonating another 1% (meaning an H+ jumps from one molecule to another leaving one with a (net) H+ charge and the other (HSO4-) with a negative charge) vs water where 1 in 10 million water molecules ionize.
In chemistry, protonation is the addition of a proton (H+) to an atom, molecule, or ion. Protonation is possibly the most fundamental chemical reaction. Usually, protonations are reversible and the conjugate base is unchanged by being protonated.
The relative static permittivity of a solvent is a relative measure of its polarity. For example, water (very polar) has a dielectric constant of 80.10 at 20 °C while n-hexane (very non-polar) has a dielectric constant of 1.89 at 20 °C.
The equilibrium
constant for the autoprotolysis (of sulfuric acid) is
Kap(25°C)= [H3SO4+][HSO4-] = 2.7×10^-4
My guess is this means about 1% of H2SO4 molecules ionize
(.01 x .01 = 10^-4).
The comparable equilibrium constant for water, Kw is 10^-14, a factor 10 billion smaller.
-- Sulfuric acid reacts with most metals via a single displacement reaction to produce hydrogen gas and the metal sulfate. Dilute H2SO4 attacks iron, aluminium, zinc, manganese, magnesium and nickel, but reactions with tin and copper require the acid to be hot and concentrated.
-- the dilute acid reacts with metals high in the reactivity series (such as Zn) to produce a salt and hydrogen. This is explained more fully in 'A New Certificate Chemistry' by Holderness and Lambert.
The equilibrium
is actually more complex than shown above; 100% H2SO4 contains the following
species at equilibrium (figures shown as millimoles per kilogram of solvent):
HSO4- (15.0), H3SO4+ (11.3), H3O+ (8.0), HS2O7- (4.4), H2S2O7 (3.6), H2O
(0.1)
-- In chemistry, polarity refers to a separation of electric charge leading to a molecule or its chemical groups having an electric dipole or multipole moment. Polar molecules interact through dipoledipole intermolecular forces and hydrogen bonds. Molecular polarity is dependent on the difference in electronegativity between atoms in a compound and the asymmetry of the compound's structure. For example, a molecule of water is polar because of the unequal sharing of its electrons in a "bent" structure, whereas methane is considered non-polar because the carbon shares the electrons with the hydrogen atoms almost uniformly. Polarity underlies a number of physical properties including surface tension, solubility, and melting- and boiling-points.
------------------------------------------------
Univ
of Sydney electrochemistry lecture
Electrochemistry
from material fromm Univ of Sydney (Australia) (link below figure)
The Sydney (introductory) battery lecture shows the cell below: zinc and copper in a single electrolyte (copper sulfate). Note the voltage (potential) curve in the figure. The voltage (potential) of the electrolyte is shown (substantially) more positive than of both electrodes. This is due to nernst potentials, a space charge layer at both electrode interfaces that block further disolution of the positive metal ions into the electrolyte. The text confirms that the electrolyte voltage more positive than both electrodes.
'Sydney lecture cell'
Cu and Zn are both negative relative to electrolyte
Zn is the negative terminal of the battery (-1.1V
relative to Cu)
Zinc acts as the metal (electronegativity = 1.65)
and copper as the non-metal (electronegativity = 1.90)
(even though Zn (#30) is to the right of Cu (#29)
on periodic table)
(source -- http://www.physics.usyd.edu.au/super/life_sciences/E/E5.pdf)
Below is a simple circuit model I drew up for the Sydney zn/cu (lecture) cell (above) with two local batteries, one for the anode reaction and one for the cathode reaction. Since the Sydney figure (above) clearly shows the electrolyte more positive than either of the electrodes (due to space charge layers as shown at the two interfaces), the batteries in my sketch are oriented with the positive sides connected together and the voltage of that node representing the voltage of the electrolyte. The Sydney lecture, and Wikipedia too, say the electrolyte voltage in a cell is not really measurable.
But let a little current (Io) flow and you can see this makes a lousy battery. If the (non-metal) Cu battery is oriented opposed to the cell voltage, as the Sydney lecture indicates it must be, then some of the power out of the zinc battery (zinc electrode reaction) flows into the copper battery and gets dissipated (as heat). The copper electrode reaction is not outputting power it is being driven. This zinc/copper cell does function as a battery, it has a cell voltage which is the difference between the zinc and copper voltages, but the efficiency of this 'battery' will be lousy. The reason is that only one electrode reaction is generating usable power and some of that power is being used internally forcing the reaction at the other electrode, which since it has power flowing into it will run hot! (This point is totally glossed over in the Sydney lecture.)
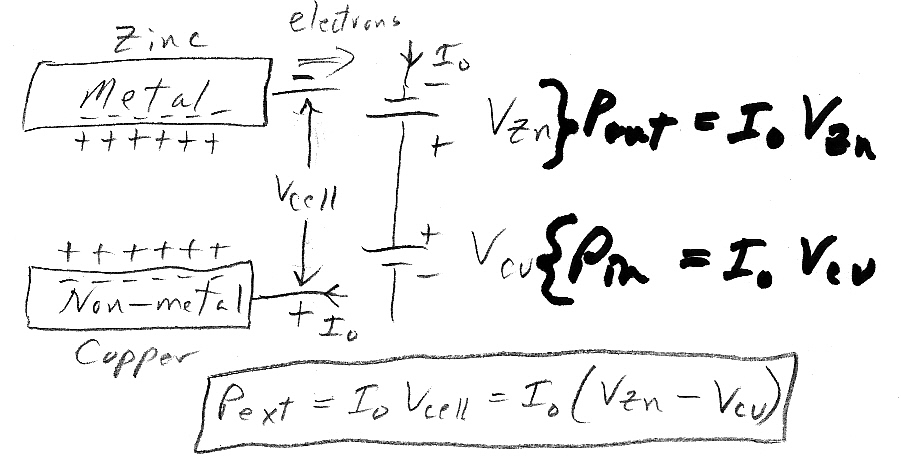
Circuit model for the 'Sydney lecture cell'
(Note some of the power from the (anode) zinc battery
is dissipated in the (cathode) copper battery,
so this is a 'battery' with very poor efficiency)
Do any 'real' batteries work this way? I doubt it. I suspect this is a simple lecture 'battery' or cell. The point Sydney makes that the electrode voltage is very misleading, because it would be natural for a student to assume that this typical of how batteries work (I initially did). If the polarity of non-metal (reduction) battery is flipped, so it points the same way as the cell voltage, which would be the case if the reduction reaction has a voltage reversed from the oxidation reaction, then the electrolyte voltage is not higher than both electrodes, its intermediate (between) the voltages of the two electrodes. I suspect in nearly all practical batteries voltages the redox voltages of the two electrode reactions have different polarities, pointing the two batteries in the same direction and causing both electrode reactions to contribute to the battery output power.
Why does the Sydney battery lecture include this misleading cell? The only thought I have is that it was the simplest cell they could draw, a combining of two of the simple electrode reactions previously discussed. But then emphasizing that the electrolyte voltage is more positive than either electrode voltage, without noting that this doesn't happen in real batteries, I find extremely misleading to someone just learning about batteries. (It certainly faked me out for a few days!)
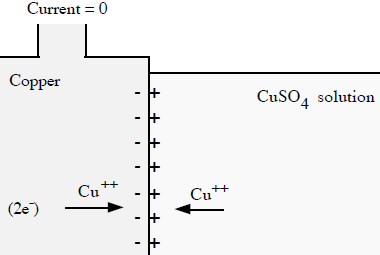
electrode: copper
electrolyte: copper sulfate
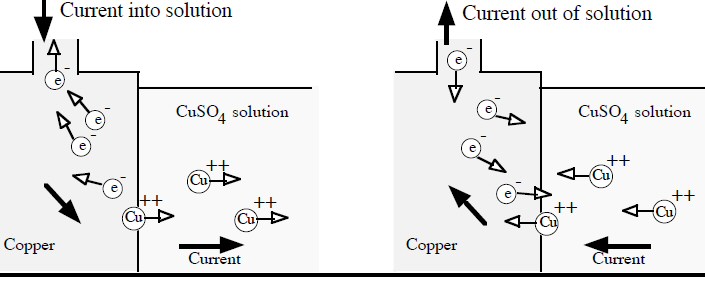
left: charge (acting as non-metal)
right: discharge (acting as non-metal)
copper ions from electrode dissolve into copper sulfate
electrolyte copper ions from electrolyte
plate onto copper electrode
electrode: copper
electrolyte: copper sulfate
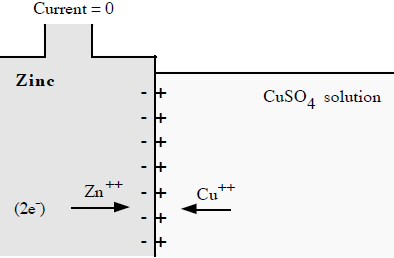
space charge build up (open circuit)
to limit Zn++ diffusion (disolving) into ionic electrolyte
electrode: zinc
electrolyte: copper sulfate
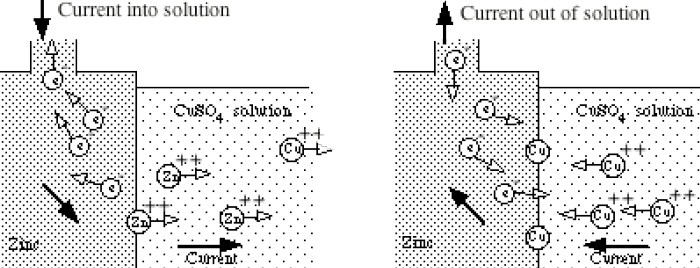
left: discharge (acting as metal)
right: charge (acting as metal)
zinc ions 'contaminate' copper sulfate electrolyte
copper ions from electrolyte plate onto zinc
electrode: zinc (metal, negative terminal)
electrolyte: copper sulfate
Concentration cell
simple 58 mv battery made by x10 difference in concentration
(58 mv = kT/q x ln(C1/C2) = 25 mv x ln(10)
is nernst voltage across porous wall to limit diffusion)
Daniell cell
----------------------------------------------
read --- details of how redox measurments are made
http://www.geochemie.uni-bremen.de/downloads/redox/chapter01.pdf
=============================================================
I saw a simple battery sciece kit in a store, so I bought it. It comes with two narrow strips of zinc and copper, and they recommend using a lemon or potato. It comes with an LCD clock as a load, and they show using two cells in series to drive IT. A zinc/copper/lemon battery is going to be a Volta battery, so if I am right about it having self discharge, then the zinc will begin to dissolve (and heat) if just put as a single electrode into the lemon. (If the chart is right and lemon ph is 2, then its H+ strength is about 1% of a strong acid (ph=0), so this is a test that the self discharge is small with weak acids.)
Here's a useful little chart I found giving the Ph of household products.

Another experiment is to mix acids and bases, which is exothermic (like lemon juice and sodium bicarbonate).
A lemon battery tutorial shows says a steel paper clip can replace zinc (yup, see below) and that voltage is about 700 mv and max current about 1 ma.
My vegtable battery tests
What, 700
mv @ 1ma? I get nothing like this. I built a potato, lemon, tap water and
tap water with salt battery all using the small zinc and copper strips
(only few mm wide) from the kit.
Open circuit
700 mv -- 950 mv
10k load
350 mv (35 ua) -- 200 mv (20 ua)
Open circuit the voltage (tested for a minute or so) was stable in 700 mv to 950 mv range. When loaded with 10k (only resistor I tried), the voltage in the potato and lemon cells dropped to 350 mv (potato) to 200 mv (lemon). This is only 35 to 20 ua of current! Tested for 15 sec or so the voltage under load would continue to drop (maybe a mv/sec). Is copper developing hydrogen bubbles?
My zinc dissolve in acid test
Left the small thin
zinc strip stuck in a lemon for 15 hours. It is a little discolored and
corroded, but no visible piting or size reduction. Who knows how much acid
was touching the zinc. Lemon ph is claimed to be 2. If all this is true,
then reactions slow way down as concentration goes down.
ph = -log (H+) where H+ (mol/L)
Are reaction rates proportional to concentration?
For lemon at ph
= 2 the H+ concentration is 0.01 mol/L, but a strong acid like in youtube
can be 10 mol/L (or higher) ??. If it took a zinc coin 1 min to dissolve
in 10 mol/L acid, then a linar scaling gives 1,000 min (16 hr). But it
seems that it's going slower than linear. It might be of course that if
stir the acid things would speed up and then look linear.
Another argument
that reactions go slow at low concentrations is that you can put your hand
in lemon juice without a problem.
-------------------------------------------------------------------------------
Water equilibrium constant
equilibrium constant defined kw = H= x OH-/H2O
where all concentrations are in mol/liter
water's mol/liter = (1000g/L)/(18g/mol) = 55.6 mol/L is taken as constant
(@25C)
Important result, which must always be true, is product
of concentration H+ x OH- = constant
(this is like in semiconductors hole x electron densities = constant)
kw
= H+ x OH- = 1 x 10^-14
So at ph=7, concentration of H+ and OH- are same at 10^-7
mol/L
-- An
aqueous solution near pH = 7 is considered neutral since [H+] = [OH-].
An acidic solution has a pH less than 7, while a basic solution has a pH
greater than 7. The conventional pH scale ranges from 0 to 14. This does
not mean the pH cannot be less zero; as an example, a solution with 10
moles per liter of H+ has a pH of -1, since [H+] = 10^+1. (ph = -log
(H+)
-------------------------------------------------------------------------------------------------
Compare to bond energy (1 ev = 23.1 kcal/mol =
96.5 kj/mol)
OH- + H+ => H2O
E0 = +?? exothermic
O-H bond energy
111 kcal per mole (4.81 ev)
student data
H+(aq) + OH-(aq) => H20(liq)
-158.23 kJ/mol (1.64 ev)
Heats of formation (1 ev = 96.5 kj/mol)
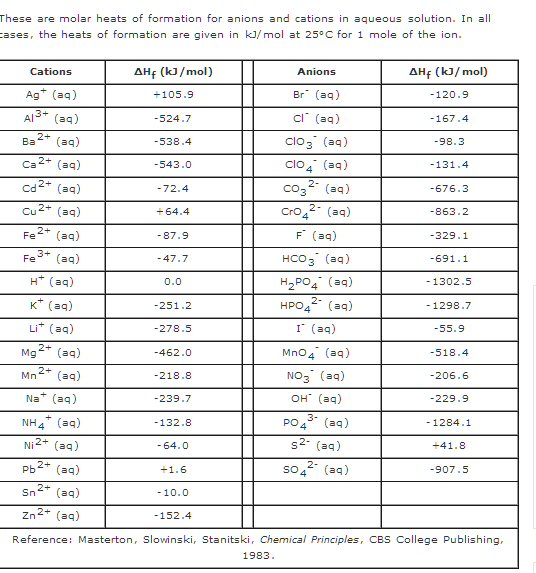
source -- http://chemistry.about.com/od/chartstables/a/heatoformions.htm
Ionic heats of formation
Seems like
the heats of formation in table above ought to be closely tied (same?)
to the standard potentials. Well they are not same, about x2 higher (1.91
to 2.08). The signs are right. Zn is negative, which is exothermic, it
wants to dissolve, while Cu is positive, it wants to come out of solution.
Zn(s)
=> Zn2+ + 2e-
Eo=0.76V
Zn2+ (above) = -152 kj/mol
-1.58 eV
Cu2+
+ 2e- => Cu(s)
Eo=0.35V
Cu2+ (above) = 64.4 kj/mol
0.68 eV
What does this all mean? It might mean, in a battery when zinc dissolves, it release approx twice the energy that is captured externally, half presumably dissipated as heat in the battery and the other half coming out electrically? But I never seen any battery info saying that half the battery energy storage gets dissipated within the batter, whichh is what this implies! Battery heat losses are always shown as primarily ohmic losses proportional to the current square drawn.
So can the heat released when a zn+ ion forms (presumably by dissolving in acid or base) be (appox) double the electrical battery power contributed by the zinc reaction?
-- The heat of formation of water from its ions (H+ + OH-) must not be confused with the heat of formation of water from its elements. (1913 textbook).
Ionization chemistry
I found in
a 1913 chemistry textbook online
some useful data.
1) Neutralization of acids by bases --- Chemists had found that when equal qantities of various strong acids (meaning acids that ionize fully) and various strong bases are diluted with water (10:1 say), and then mixed, the acids and bases neutralize forming water and heat comes out. The interesting thing is that the measured heat that comes out is always the same. The explanation is that the ions on both sides of the equation (except for H+ and OH- that merge to form water) don't react. This shows that in acids, bases, and in the neutralized product all the ions (except H+ and OH-) are separate, and all the heat must be from H+ and OH- merging to form H2O. (who knows what the units are below in this 100 year old data).
HC1 + NaOH => NaCl + H2O
13.75 CaL
HC1 + KOH => KC1 + H2O
13.70 CaL
HC1 + LiOH => LiCl + H2O
13.70 CaL
HN03 + NaOH => NaN03 + H2O
13.68 CaL
HN03 + KOH => KNO3 + H2O
13.77 CaL
2) Thermoneutrality
of Salt Solutions --- When two dilute salt solutions are mixed, there is
neither evolution nor absorption of heat. For example,
when dilute solutions of sodium nitrate and potassium chloride are mixed,
there is no thermal effect. A non-ionic equation shows a reaction (might
be happening) with sodium chloride and potassium nitrate forming (in liquid).
NaN03 + KC1 = NaCl + KNO3 0 Cal
But an ionic equation, if all the salts are fully ionized, shows no reaction (same ions on both sides). No reaction means no heat in or out.
Na+ NO3- + K+ + Cl- => Na+ Cl- + K+ + NO3-
NaCl
Heat of formation
of sodium chloride (aq) is given by Wikipedia as 407 kj/mol. The above
table shows
Na+ (aq)
-239.7 kj/mol
Cl-(aq)
-167.4 kj/mol
------------------------
-407.1 kj/mol (agrees exactly with heat of formation
of NaCl. This is an exothermic reaction)
So I find the 'Heat of formation' of an ionic solid (NaCL) is the same of the sum of the 'Heats of formation' of its two ions in water (Na+ and Cl-). How is the energy divided? By atomic weight? By kinetic energy? Atomic weight of sodium is 23 and chlorine 35.5. If I assume no momentum change when NaCl separates, then the lighter atom (sodium) ends up with higher velocity and higher kinetic energy by the ratio of the masses [35.5/23 = 1.54]. This is approx what happens, since the 'heat of formation' energy of sodium ion is x1.43 the energy of the chlorine ion.
Water Liquid H2O -285.83
Silver Oxide Solid Ag2O
-31.1
ZnO(s)
-348.0
Mercury(II) Oxide (red) Solid HgO
-90.83
Aluminum Oxide Solid Al2O3
-1,675.7
Magnesium Oxide Solid MgO
-601.24
Manganese(II) oxide Solid MnO
-384.9
Manganese(IV) Oxide Solid MnO2
-519.7
Sulfuric acid Liquid H2SO4
-814
Sodium chloride Aqueous NaCl -
407.27
Sodium Hydroxide Aqueous NaOH
-469.15
Magnesium hydroxide Aqueous Mg(OH)2 -926.8
Ammonia Aqueous NH3
-80.8
Ammonia Gas NH3
-45.90
CO2(g)
-393.5
Monoatomic oxygen Gas O
249
Chlorine Gas Cl2
0
Copper Solid Cu
0
Hydrogen Gas H2
0
http://chemistry.about.com/library/weekly/blheatform.htm
http://en.wikipedia.org/wiki/Standard_enthalpy_change_of_formation_(data_table)
http://en.wikipedia.org/wiki/Standard_enthalpy_of_formation
-- Heat of Formation (Wikipedia) --- The standard enthalpy of formation or standard heat of formation of a compound is the change of enthalpy that accompanies the formation of 1 mole of a substance in its standard state from its constituent elements in their standard states (the most stable form of the element at 1 bar of pressure and the specified temperature.)
-- All elements in their standard states (oxygen gas, solid carbon in the form of graphite, etc.) have a standard enthalpy of formation of zero, as there is no change involved in their formation.
-- Standard enthalpy of solution (or enthalpy change of dissolution or heat of solution) is the enthalpy change associated with the dissolution of a substance in a solvent at constant pressure under standard conditions, as previously defined.
-- Standard enthalpy of neutralization is the change in enthalpy that occurs when an acid and base undergo a neutralization reaction to form one mole of water under standard conditions, as previously defined. The standard enthalpy change of neutralization for a strong acid and base is -57.3 kJ/mol.
-- Just as the energy of forming a chemical bond is the difference between electron affinity and ionization energy, the heat of solution of a substance is defined as the sum of the energy absorbed, or endothermic energy, expressed in positive values and unit kJ/mol, and energy released, or exothermic energy (negative value).
-- The energy involved in removing electrons to make a cation is called the ionization energy. The enthalpy of adding electrons to an atom to make it an anion is called the electron affinity.
-- Ionization energy --- In chemistry, the value is usually given in kJ/mol (or formerly kcal/mol). This value is strictly the "molar ionization energy" and corresponds to the energy required to remove (to infinity) one mole of electrons from one mole of gaseous atoms or molecules. However it is often just called "ionization energy". Na ionization energy = 496 kj/mol
-- Dissolution
can be viewed as occurring in three steps:
1) Breaking solute-solute attractions (endothermic), see for instance lattice
energy in salts.
2) Breaking solvent-solvent attractions (endothermic), for instance that
of hydrogen bonding
3) Forming solvent-solute attractions (exothermic), in solvation.
-- The lattice energy of an ionic solid is a measure of the strength of bonds in that ionic compound. It is usually defined as the enthalpy of formation of the ionic compound from gaseous ions and as such is invariably exothermic.
Latice energy NaCl -787 kJ/mol
-- In weak basic solutions containing Zn2+ ions, the hydroxide Zn(OH)2 forms as a white precipitate. In stronger alkaline solutions, this hydroxide is dissolved to form zincates ([Zn(OH)4]2-
Measuring
standard reduction potential
All the descriptions
I read of how 'standard reduction potentials' are measured are unclear.
No one gives a circuit! It never occurs to anyone to show where the reference
point is for the voltage being measured! And an obvious measurement problem
that I have yet to see addressed is that if the reaction is not spontaneous,
say reduction of zinc, then it must be driven. So I have made two sketches
showing how, at least in principle, 'standard reduction voltage' can be
measured.
In the circuit below the metal being measured is at the top and the hydrogen reference electrode at the bottom. The adjacent electrolytes have the metal and H+ ions at 1.0 mol and between them is a high impedance salt bridge (ion resistor). I have grounded the hydrogen terminal. {Functionally where the ground is makes no difference, it simply provides a reference point for the measured metal electrode voltage or (Vs) }. The metal terminal voltage, measured relative to the hydrogen gnd, is the 'standard reduction voltage' of the tables. What I have added, and have not seen mentioned anywhere, is that the metal terminal voltage is forced by a supply (Vs), which can go positive and negative.
The measurement process is to vary Vs to find the point where current (I) goes to zero. This voltage is the 'standard reduction voltage' of the tables. Current is zero when Vs has the same polarity and amplitude as the space charge voltage (local battery) at the metal electrode interface. A small variation in Vs above and below this (current null) point reverses the direction of current (I) flow, so the reaction at the metal electrode must switch between oxidation and reduction, and the direction of ion flow in the electrolyte must reverse.
Zinc spontaneously 'wants' to dissolve so that makes it's space charge E field point toward the zinc (upward) with the current null point for zinc [Vs = -0.76V]. Copper ions in solution wants to plate out on the copper electrode, so copper's space charge layer E field points away from the copper (download) with the current null point [Vs = +0.35V]. These voltages are the 'standard reduction voltages' found in the tables for zinc and copper ionizing.
my sketch for measuring 'standard reduction potential'
(Vs where I = 0)
(ajacent solution have metal ion or H+ at 1.0 mol)
The sketch below shows what I think the current would be (for zinc alone and copper alone) as Vs varies. The straight line slope (delta I vs delta Vs) is detemined by the resistance of the salt bridge. For positive I (see sketch above) the electron flow is outward, so the metal reaction is oxidation, and for negative I it is reduction. This sketch shows that if the terminals are shorted (Vs=0V), zinc battery action produces positive I, which is outward electron flow and oxidation. For (Vs=0V) with copper its battery action cause negative I, which is inward electron flow and reduction. I dotted the non-battery range of each electrode only because I don't know how well they take to being driven (but I suspect the line remains straight).
How I expect current (I) would vary with Vs in circuit
above
(positive I is defined in circuit sketch)
zinc (left) has a current null at Vs=-0.76V
copper (right) has a current null at Vs=+0.35V
After writing above, I found in a chaper from a book online about measuring redox in the water supply the redox measuring circuit (below). Actually its for measuring the redox potential of a 'redox pairs', which are either ions that can exist in solution in two ionized states, or solids or gases that ionize.
source --- http://www.geochemie.uni-bremen.de/downloads/redox/chapter01.pdf
Notice how sloppy this diagram is. The polarity of the measured voltage is not indicated either in the figure or in the text below. All that would be necessary is to put +/- signs on the voltmeter, or to show a reference ground. The figure does neither.In natural water supplies the redox pairs of interest are in Eo order (+800 mv to - 400 mv): O2/H2O, NO3-/N2, MnO2(s)/Mn2+, Fe(OH3)(s)/Fe2+, SO4 2-/H2S, CO2/CH4, H+/H2. This is in natural, outside water supplies where Ph is about 7, so H+ and OH- concentrations are 10^-7, thus all the redox voltages are shifted negative by about 400 mv. H+/H2 , which is at zero at standard conditions here is at the bottom is about - 400 mv. O2/H2O is here near +800 mv, which at standard conditions would put it about +1.2V, which is the voltage for electrolsis of water.
The first thing that puzzles me is the redox pair voltage range: 1.2V (+800 mv to -400mv). This is double the standard redox voltage range for metal ionizing which is 600 mv (+300 mv to -300 mv at standard conditions).
Ionic bonding
Ionic bonding
is one of the keys to batteries, so I have been trying to understand ionic
bonds. The key mystery, and an apparent contradiction, is how on the one
hand is it that the ionic bond is so strong, demonstrated by the high melting
point of salts (NaCl melts at 800C), yet when ionic solids dissolve in
electrolytes (or melt) the ions are free and they conduct current well.
How come these strong ionic bonds in (polar) liquids just come apart?
The bond in a salt like NaCl is only a few percent covalent (shared), so it's a pretty good approximation to say that each atom really is a charged ion, that the sodium extra electrons have jumped to chlorine. In the NaCl crystal (see figure) each node is not an NaCl pair, it's an ion (Na+ or Cl-). Each ion
I am coming to realize that it is very misleading to compare bonds by considering the attraction between two atoms. A covalent bond (H2, O2, N2, Cl2) does pull two atoms together into a molecule, but the electron sharing produces basically only a pull toward each other. A key point is that because of the symmetry of the molecule there is no significant net charge visible from far away, and the result is no significant attraction between molecules. That's why H2, O2, N2, Cl2 are all gases!Thinking of a two atom ionic molecule maybe provides a (qualitative) picture for how they dissolve. Any exposed 'dumbbell' end in a polar liquid will be pulled on by the charge of the liquid molecules. And since I remember reading dielectric constant is a measure of how large a charge dipole is, and since the dielectric constant of water is high, it is quite a strong polar liquid and hence good dissolver of ionic crystals.In contrast if a two atom 'crystal' of NaCl is considered it's basically a charge dumbbell. It couldn't have a stronger charge as viewed from outside. Such a molecule would pull like crazy on every other ion, that's why NaCl auickly assembles into huge 3d grids of alternating + and - ions.
The other obvious question is why don't ions in liquid stick to each other and regrow a crystal. Quantatively this must be indicated by a negative Eo for the equation below. In other words is must not be thermodynamically favorable to regrow the crystal. Of course, this doesn't provide a picture of why not. It must somehow be that the pull/jostling of the moving liquid charges are able to break up any two NaCl ion pairs that form.
Na+ + Cl- => NaCl Eo must be negative
Well, as gases sodium ions and chlorine ions really want to come together to make solid sodium chloride. This is a very exothermic reaction (see figure below). In fact 787 kj is identified by Purdue chemistry lecture notes as the lattice energy of solid sodium chloride. But apparently the situation must be very different for Na+ and Cl- in water.
Na+(g) + Cl-(g) +> NaCl(s) Ho = -787.3 kj x (1 ev/96 kj) = -8.2 eV
Ionic bonding notes
I checked out the
two types of strong bonds between atoms in Wikipedia: covalent bonds and
ionic bonds. The 'classic' covalent bond is between two identical atoms,
like hydrogen. They come close enough so that their outer (valence) electron
orbitals overlap as shown below. Covalent bonding is described at the atoms
sharing
valence electrons.
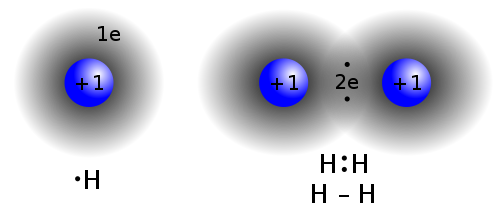
The classic ionic bond holds salt (NaCl) together: sodium (group 1 metal) with chlorine (group 17 non-metal). The animated figure below clearly shows an electron jumping from sodium (some energy is needed to ionize) to chlorine (some energy is output). The two oppositely charged ions then move closer together, but the orbits are not shown overlapping, outputting (substantially) more energy. Ionic bonding is described as one atom losing an electron and the other gaining it forming oppositely charge ions that attract.
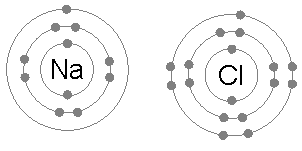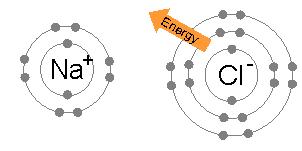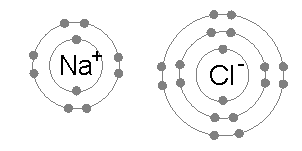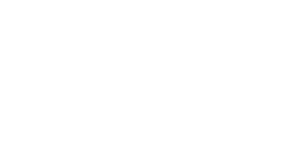
(source = tutorvista)
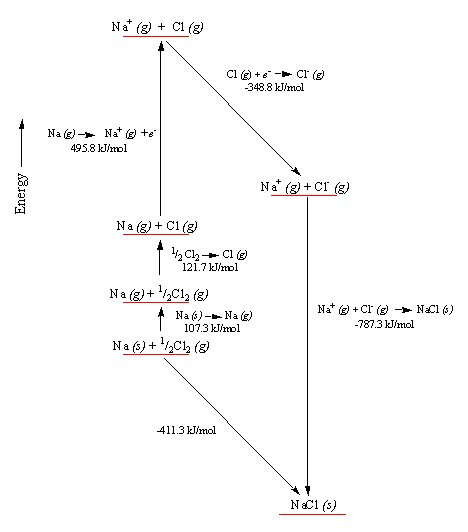
Detail steps: Na(s) and Cl2(g) => NaCl(s)
(413 kJ/mol)
Na+ + Cl- => NaCl(s) (787 kJ/mol exothermic)
endothermic: Na solid to Na gas (107),breaking Cl2
to Cl (122), removing electron from Na to form Na+ (496)
exothermic: adding electron to Cl to form Cl- (349),
Na+ joining Cl- (787)
(source -- http://chemed.chem.purdue.edu/genchem/topicreview/bp/ch7/whydoes.html)
Then (in Wikipedia 'ionic bond') you find this --- "Pure ionic bonding is not known to exist. All ionic compounds have a degree of covalent bonding." Translation: All ionic and covalent bonds (to some extent) have their valence electron orbitals overlapping. It's a matter of degree. In covalent bonds they overlap more and in ionic bonds they overlap less.
-- All ionic bonds have some covalent character. For example, NaCl and MgO bonds have a few percent covalency, while SiO bonds are usually ~50% ionic and ~50% covalent. Predominantly covalent bonds with partial ionic character are called polar covalent.
-- Ionic compounds consist of ions and not molecules (Note, each node in the crystal is an ion).
-- Ionic compounds have high melting and boiling points. For example, sodium chloride has a high melting point of 1,472 F (800 C) and boiling point of 2,575 F. Lithium chloride has a melting point of 1,122 F and boiling point of 2,462 F. Potassium chloride has a melting point of 1,526 F and boiling point of 2,732 F. As these compounds contain ions held together by strong electrostatic forces, very high amount of energy is required to overcome this force and break the crystal lattice. This explains the high melting point and boiling point of ionic compounds.
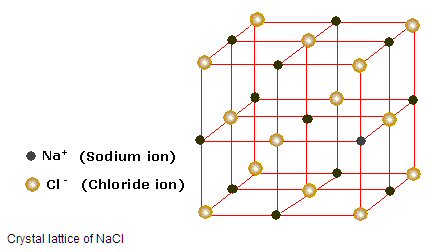
grid of 9 x 3 =27
center (blk) is attracted by 6 yel in faces + 8 yel
in corners
and repelled by 12 blk diagonals
(source = tutorvista)
Assume each line = 1 and attraction goes down as 1/distance^2
6 x face attact
6 x 1 =6
8 x corner attract
8 x (1/sqrt{3})^2 = 2.67
12 x diagonal
12 s (1/sqrt{2})^2 = 3
The diagonal repelling force is only about 1/3rd of the attractive force, and about 70% of attractive force it toward the six closest nodes in the center of sides of the cube surrounding each node.
--- Ionic compounds conduct electricity when dissolved in water. They also conduct electricity when melted. When dissolved in water, the ions are separated. The (polar) water molecules break the strong electrostatic force of attraction. The strong electrostatic force of attraction is broken while melting resulting in the formation of free ions.
*** Detail description of how water dissolves ionic materials
-- Water has
a high dielectric constant. Water easily breaks the strong electrostatic
force of attraction. The water molecule is polar in nature. The positively
charged hydrogen atoms surround the anion. The negatively charged OH- (hydroxyl)
surround the cation. Hence, the cation is separated from the anion, breaking
the crystal lattice. The organic solvents contain non-polar molecules.
Therefore, they are unable to break the electrostatic forces of attraction.
(source -- http://www.tutorvista.com/content/chemistry/chemistry-i/chemical-bonding/ionic-compounds.php)
How polar is water?
One fairly
recently did I learn that the dielectric constant (epsilon or e, or permittivity)
of a liquid is a (good) measure of how polar it is. I know a lot about
'e' (as applied to solids) because it's it's a key parameter of capacitors,
a measure of how much energy the capacitor can store in the electrical
field. Energy stored in a vacuum has an energy density of [E = (1/2) eo
E^2]. Energy stored with the same E feld in the same region of space
containing a material is [E = (1/2) e E^2]. The energy storage goes
up by the ratio of e/e0, and this ratio is known as the relative permittivity
or dielectric constant.
For water e/eo = 80, a high value. The polar nature of the water molecule increases the E field energy storage by a factor of 80 over a vacuum, and higher by a factor of 40 compared to non-polar liquids that can have a permitivity of 2 like hexane, a linear carbon chain dotted with hydrogen. (see Wikipedia 'Relative permittivity')
salt(s)
3 - 15
water
80
sulfuric acid
84
An E field applied to a material exerts forces on the atoms' electron orbitals (& maybe ions), which have a spring like character. A DC E field in effect pulls on all these springs (the picture is it slightly distorts the orbitals) storing a little energy in each atomic 'spring', then when the electron 'springs' relax the energy is released. This is how most of the energy in capactors is stored.
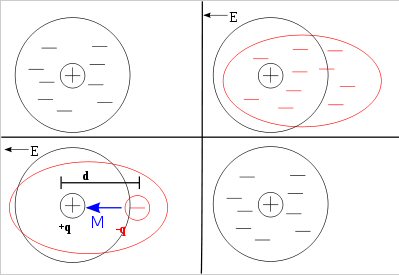
Classic atomic dielectric model showing E field distorting
an atom's electron 'cloud',
and generating a dipole moment (M)
(source -- Wikipedia 'Dielectric')
E field energy storage in a liquid
But how does
an E field affect a liquid with polar molecules? Thinking about it I suspect
mechanism for E field energy storage in a liquid may be very different
than in a solid. In a liquid the molecules can move. My guess is the 'spring'
mechanism when an E field is applied to a liquid with polar molecules is
that the molecules partly align with the E field fighting opposing the
themal forces that would random align them and probably rotate them too.
Yup, this seems to be basically what Wikipedia says (below), and polarization of (liquid) polar molecules has a special name, 'dipolar polarization'.
"If a dielectric is composed of weakly bonded molecules, those molecules not only become polarized, but also reorient so that their symmetry axis aligns to the field."This picture is consistent (maybe!) with the curve below that shows a large roll off (90 to 20) in permittivity of water as temperature rises. My explanation of this is that as the randomizing thermal forces get stronger the E field is less and less able to align the molecules, (but it's not quite obvious to me that this leads to less energy storage, which is what permitivity is all about).** "Dipolar polarization is a polarization that is particular to polar molecules. This polarization results from permanent dipoles, e.g. asymmetric bonds between oxygen and hydrogen atoms, which retain polarization in the absence of an external electric field. The assembly of these dipoles forms a macroscopic polarization."
source -- Wikipedia 'Relative permittivity'
Ion size
In reading about ions
and trying to understand more about how ions form the issue of ion size
(ionic radius) came up, and here I stumbled onto the a minor mystery.
Some references claim that non-metals that ionize to negative ions by adding electron(s) into 'holes' in their nearly complete outer shells, like chlorine, expand (substantially) in radius as they ionize, as shown in this textbook sketch below.

Textbook sketch showing Na shrinks as it ionizes
and (surprisingly) Cl expands as it ionizes
(source -- http://www.chemguide.co.uk/atoms/properties/atradius.html)
Others references say the swelling of chlorine when it ionizes is misleading pointing out that it depends on the baseline radius: covalent radius or van de wall radius. An atom's covalent radius is smaller because it's half the atom to atom spacing in a molecule like Cl2, where the orbitals are squished (overlap quite a bit). The van de wall radius is half the distance between two atoms loosely bonded, and hence can be considered to be (approx) the radius of an isolated atoms.
Ionic solids consist of an ion matrix
The effective
radius of the sodium atoms shrinks, because when ionized to Na+ is loses
the only electron it has in the 3rd electron shell. Below shows Na+ radius
is only 102 pm much smaller than all of sodium atom's other radii. On the
other hand the radius of Cl- is much larger than the chlorine atoms
atomic or covalent radius. (I don't know how atomic radius is defined.)
A confirming number for the ionic radii of these two atoms is the lattice
constant of crystal (NaCl) salt, which is the dimension of a lattice 'cell',
which must mean a (repeating) Na+, Cl- pair. (All dimensions below in pm.)
atomic covalent
van de wall ionic radius
---------- -----------
------------- --------------
Sodium
186
166
227
102 (Na+)
Chlorine
99
102
175
181 (Cl-)
---------------------
total 283 NaCl
(1/2) NaCL (salt lattice constant, cell dimension) (1/2) x 564 = 282It looks very surprising that chlorine would swell up (almost by a factor of two) when it picks up one electron to fill the hole in it outer shell. I had assumed that its radius would basically not change. Wikipedia's explanation (for similar fluorine) is "The ionic radius of fluoride is much larger than its covalent radius. When F becomes F-, it gains one electron but has the same number of protons, meaning the attraction of the protons to the electrons is weaker, creating a larger radius."
Calculated NaCl spacing from densityChlorine ion expands? --- wrong baseline
The spacing between Na and Cl atoms in a salt crystal should be calculable from the density of a salt crystal. Since along x, y, and z Na and Cl ions alternate the spacing between every pair of ions should be the sum of a sodium ion radius and chlorine ion radius (as they are bonded in NaCl. So for fun I did the calculation.The result (I thought) should be the value above 283 pm, but when I did the calculation (below) I got 355 pm. This is high by x1.256 and suspiciously when cubed it's almost exactly x2. Below I assume a mole of salt had 6.02 x 10^ 23 ions, but does maybe a mole of salt have 6.02 x 10^23 (sodium + chlorine ion pairs)? If a substance is made of molecules, then a mole of it is 6.02 x 10^23 molecules, but ionic substances like salt do not form molecules (as one chemistry reference emphasized), its crystal grid consists of alternating Na+ and Cl- ions.
salt density 2.16 x 10^3 kg/m^3
Na atomic weight 23
Cl atomic weight 35.5
Avagardo's number 6.02 x 10^23Vol of mole of salt = (23 + 35.5 grams)/2.16 x 10^6 gram/m^3
= 27.1 x 10^-6 m^3
Linear dimension
of NaCL mole = cube root{27.1 x 10^-6 m^3}
3.00 x 10^-2 m (cube 3 cm each side)
Cube root of
Avagardo's number = cube root{602 x 10^21}
= 8.44 x 10^7Salt ion-ion
spacing? = 3.00 x 10^-2 m/8.44 x 10^7 atoms
= 0.355 x 10^9 m (355 pm)Explanation
However, if the 58.5 gram, 30 mm cube mole of salt has not Avagado's number of ions but Avagardo's number of ion pairs (Na-Cl pairs), (even though there is no obvious pairing in the crystal!), then the number of ions in the volume of the sample is doubled so the ion-ion spacing reduces by cube root{2} making it 282 pm, which agrees almost exactly with the tabulated values.Better explanation
After thinking about this, I see a better way to get the 'right' answer. It is true that assuming a 55.5 gram mole sample of salt contains 6.02 x 10^23 ion pairs, or x2 Avagardo's number of nodes or ions, does give the right ion-ion spacing, but it seems suspect.Later it hit me I can get the same answer by redefining how a mole is calculated for an ionic solid. Instead of summing the atomic weights of the two atoms I should use the average molecular weight. This cuts the volume of a mole of salt in half. The nodes in a simple ionic crystal, like salt, alternate back and forth between its two ions, so it makes some sense to use the average ion weight when calculating the grams of a mole. With this new definition a mole of salt now weights [(23 +35.5)/2] = 27.75 grams (rather than 55.5 grams), so Avagardo number of ions (nodes) in this (half) volume gives the right ion-ion spacing. This is much cleaner.
It is true that the Cl- ion radius is much larger than the chlorine atom has when it is covalently bonded in chlorine gas (Cl2), so from this point of view it expands when it ionizes. But from another point of view what happens when it ionizes is just that it 'unsquishes', i.e. it rounds out to the radius of an isolated chlorine atom, which is approximated by the van de wall's radius. So from this point of view, with this baseline, chlorine does not expand as it ionizes to Cl- with an electron popping into its one vacant opening in the 3rd shell, consistent with common sense!
Below notes from Jim Clark of ChemGuide (for link see figure above), retired UK chemistry instructor and author of several books. An excellent reference, because his aim is to make clear at HS level and beginning college level chemistry points that chemistry books do not explain well. On the swelling negative ion problem he says he was challenged by a senior professor that the standard (electrons not pulled so hard) explanation did not make sense, and he worked on it for a week and talked to a lot of experts.
left -- (squashed) covalent or metallic radius (102 pm for chlorine)
right -- (just touching) van der waals radius (175 pm for chlorine)
(source -- http://www.chemguide.co.uk/atoms/properties/atradius.html)
-------------------------------------------------------------------------------------------- (re: figure) The type of atomic radius being measured here is called the metallic radius or the covalent radius depending on the bonding. The right hand diagram shows what happens if the atoms are just touching. The attractive forces are much less, and the atoms are essentially "unsquashed". This measure of atomic radius is called the van der Waals radius after the weak attractions present in this situation.-- It is perfectly true that negative ions have radii which are significantly bigger than the covalent radius of the atom in question. And the argument then goes that the reason for this is that if you add one or more extra electrons to the atom, inter-electron repulsions cause the atom to expand. Therefore the negative ion is bigger than the atom.
-- This seems to me to be completely inconsistent. If you add one or more extra electrons to the atom, you aren't adding them to a covalently bound atom. You can't simply add electrons to a covalently-bound chlorine atom, for example - chlorine's existing electrons have reorganized themselves into new molecular orbitals which bind the atoms together.
-- In a covalently-bound atom, there is simply no room to add extra electrons. So if you want to use the electron repulsion explanation, the implication is that you are adding the extra electrons to a raw atom with a simple uncombined electron arrangement.
-- In other words, if you were talking about, say, chlorine, you are adding an extra electron to chlorine with a configuration of 2,8,7 - not to covalently bound chlorine atoms in which the arrangement of the electrons has been altered by sharing.
-- That means that the comparison that you ought to be making isn't with the shortened covalent radius, but with the much larger van der Waals radius -- the only available measure of the radius of an uncombined atom. Starting from the van der Waals radius, which is the radius of a bare (unsquashed orbitals) chlorine's ionic radius just grows slightly 181 vs 175.
--- It seems to me (Clark) that, for negative ions, it is completely illogical to compare ionic radii with covalent radii if you want to use the electron repulsion explanation.
Source -- http://www.webelements.com/ (search out 'Reduction Potentials' and scroll down)
How to read
Oxidation
state in green
Reduction
potential (eV) in red --- reduction is left => right (oxidation right =>
left)
Spontaneous
reduction ---positive (red) eV (example +.34V
cu2+ + 2e- => cu in acid)
Spontaneous
oxidation --- negative (red) eV (example -.79V
zn => zn2+ + 2e- in acid)
Metal oxidation overview
Most metals
with their sea of electrons want to dissolve in acids as positive ions.
These are anode metals. Only a relatively few (oddball) metals prefer to
reduce rather than oxidize (in acid) and have negative oxidation voltages
with platinium and gold being the least reactive. The oddball metals include
copper, silver, and mercury, all of which are used as cathode materials.
Li(s) => Li+ + e-
Eo=+3.04V
Mg(s) => Mg2+ + 2e- Eo
= +2.37V
Al(s) => Al3+ + 3e-
Eo = +1.66V
Mn(s) => Mn2+ + 2e- Eo
= +1.18V
Zn(s) => Zn2+ + 2e-
Eo = +0.76V
Fe(s) => Fe2+ + 2e-
Eo = +0.44V
Sn(s) => Sn2+ + 2e-
Eo = +0.137V
Pb(s) => Pb2+ + 2e-
Eo = +0.125V
Fe(s) => Fe3+ + 3e-
Eo = +0.04V
----------------
Bi(s) => Bi3+ + 3e-
Eo= -0.32V
Cu(s) => Cu2+ + 2e-
Eo = -0.34V
Ag(s) => Ag+ + e-
Eo = -0.80V
Hg(s) => Hg+ + e-
Eo = -0.80V
Pt(s) => Pt2+ + 2e-
Eo = -1.18V (unreactive, does not ionize easily)
Au(s) => Au3+ + 3e-
Eo= -1.52V
Daniell cell --- basic zinc and copper battery
Anode
zn => zn2+ + 2e- Eo=0.79V
Cathode
cu2+ + 2e- => cu
Eo=0.34V
. 
Tin
Anode
sn => sn2+ + 2e- Eo=0.137V
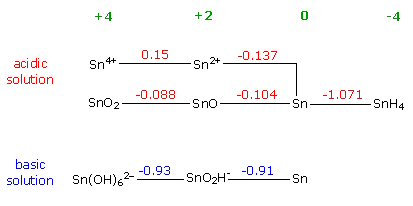
Silver and mercury
.
Salt -- chlorine and sodium
Cl2 + 2e- => 2Cl-
Eo= 1.36V
Na => Na+ + e-
Eo= 2.713V
 .
. 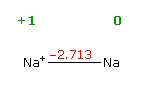
Flourine and sulfur
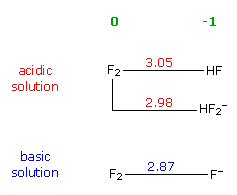 .
. 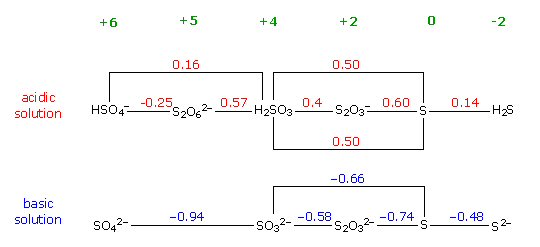
Lithium & Potassium (group 1)
Lithium is
the most reactive of all anode metals with an oxidation voltage of Eo=3.04V,
so lithium batteries tend to have the highest cell voltages in the battery
world.
anode
Li => Li+ + e- Eo=3.04V
anode
K => K+ + e- Eo=2.93V
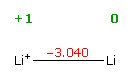 .
. 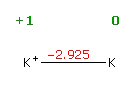
Magnesium & Calcium (group 2)
Why is calcium
not used for anodes?
anode Mg => Mg2+ + 2e-
Eo=2.36V
anode Ca => Ca2+ + 2 e-
Eo=2.84V
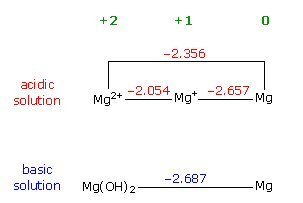 .
. 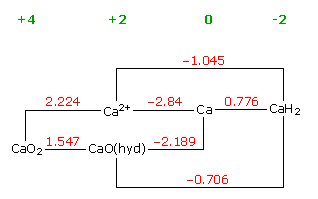
Aluminum
Aluminum makes
a good anode metal with an oxidation voltage of Eo=1.68V. (Aluminum batteries
do exist)
Oxygen
Oxygen reduced to water (in
acid)
2O2 + 4H+ + 4e- => 2H2O
Eo=1.23V
Oxygen reducted to OH- (in
base)
O2 + 2H2O + 4e- => 4OH-
Eo=0.40V
Manganese oxide
with an open crystal structure is used as the cathode material in the lithium
ion batteries used in the Volt and Leaf electric cars. Lithium ions are
able to squeeze in/out (intercalate and deintercalate) between the layers
of certain metal oxides. When the battery discharges, the oxidation state
of the manganese is reduced (MnO4 => MnO2?) and Li+ ions enter the crystal.
Can't the find the Eo of this reaction, but it probably adds about 0.7V
to the cell voltage. (Note below shows the MnO4 => MnO2 reduction voltage
is positive 1.70V for acid and 0.60V for basic, so it's probably somewhere
in this range for the lithium salt electrolyte.) When the battery is recharged,
everything reverses, the oxidation level of the manganese oxide increases
(MnO2 => MnO4?) and the lithium is driven out.
Lead
.
Liquid metal battery
As Don Sadoway's liquid
metal battery discharges the magnesium and antimony electrodes both dissolve
as positive ions from the anode and negative ions from the cathode, which
join in a salt electrolyte to make (presumably) ionic salt molecules. According
to Sadoway's patent the expected cell voltage is about 1V, so since the
magnesium reaction is stongly exothermic, it seems to imply that to force
antimony to act as a non-metal and be reduced and to dissolve it must be
driven.
Anode
Mg => Mg2+ + 2e-
Eo=2.4V
Cathode Sb + 3e-
=> Sb3-
Eo=-1.4V??
Iron
Iron (in acidic
seawater) has a fairly stong tendency to oxidize (corrode) to Fe2+ ions
with an oxidation potential of Eo=0.44, which fortunately is less than
zinc (Eo=0.76V). (It also will weakly oxidize to Fe3+ ions with an oxidation
potential of Eo=0.04V.)
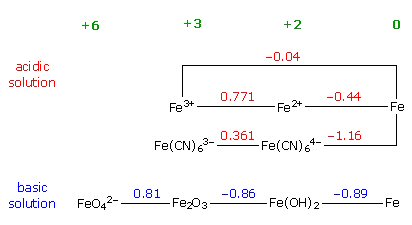
Platinium
Platninum
(Pt) is consider unreactive and often used in the lab when an unreactive
electrode is needed. It's oxidation voltage is a fairly high negative value
(Pt => Pt2+ + 2e- Eo=-1.19V), so it won't ionize in acid. Comon
anode metals like zinc, aluminum, lithium all have positive oxidiation
voltages and release positive ions. Copper has a negative oxidation voltage,
but it is only -0.34V compared to -1.19V for platinum.